
- Общая характеристика
- МНН
- Описание
- Состав
- Форма выпуска
- Фармакотерапевтическая группа
- Фармакологические свойства
- Показания к применению
- Способ применения и дозировка
- Побочные действия
- Противопоказания
- Передозировка
- Меры предосторожности
- Дети
- Период беременности
- Управление автомобилем
- Взаимодействие
- Условия и срок хранения
- Упаковка
- Правила отпуска
- Информация о производителе
Внимание! Срок действия регистрационного удостоверения 10574/17/19/20 закончился 28.06.2022
Нинларо инструкция по применению
Официальная инструкция лекарственного препарата Нинларо капсулы 2,3мг, 3мг, 4мг. Описание и применение Ninlaro, аналоги и отзывы. Инструкция Нинларо капсулы утвержденная компанией производителем.
Общая характеристика
Международное непатентованное наименование
Ixazomib.
Описание
2,3 мг: твердые желатиновые капсулы № 4, корпус капсулы светло-розового цвета, крышечка капсулы светло-розового цвета, с надписями «Takeda» на крышечке капсулы и «2.3 mg» на корпусе капсулы, нанесенными черными чернилами;
3 мг: твердые желатиновые капсулы № 4, корпус капсулы светло-серого цвета, крышечка капсулы светло-серого цвета, с надписями «Takeda» на крышечке капсулы и «3 mg» на корпусе капсулы, нанесенными черными чернилами;
4 мг: твердые желатиновые капсулы № 3, корпус капсулы светло-оранжевого цвета, крышечка капсулы светло-оранжевого цвета, с надписями «Takeda» на крышечке капсулы и «4 mg» на корпусе капсулы, нанесенными черными чернилами.
Состав лекарственного средства
Одна капсула содержит:
Активное вещество
2,3 мг: иксазомиба цитрат 3,29 мг соответствует иксазомибу 2,3 мг.
3 мг: иксазомиба цитрат 4,3 мг соответствует иксазомибу 3 мг.
4 мг: иксазомиба цитрат 5,7 мг соответствует иксазомибу 4 мг.
Перечень вспомогательных веществ
Целлюлоза микрокристаллическая
Магния стеарат
Тальк
Оболочка капсулы
Желатин
Титана диоксид (Е171)
мг: железа оксид красный (Е172)
мг: железа оксид черный (Е172)
мг: железа оксид желтый (Е172) и железа оксид красный (Е172)
Чернила
Шеллак
Пропиленгликоль
Калия гидроксид
Железа оксид черный (Е172)
Несовместимость
Не применимо.
Форма выпуска
Капсулы.
Фармакотерапевтическая группа
Противоопухолевые средства, прочие противоопухолевые средства.
Код АТХ: L01XX50.
Фармакологические свойства
Фармакодинамика
Механизм действия
Иксазомиба цитрат, неактивная форма лекарственного средства, является веществом, которое быстро гидролизуется в физиологических условиях в свою биологически активную форму, иксазомиб.
Иксазомиб является пероральным, высокоселективным и обратимым ингибитором протеасом. Иксазомиб преимущественно связывается и ингибирует химотрипсинподобную активность бета-5 субъединицы протеасомы 20S.
Иксазомиб вызывал апоптоз культивируемых invitro клеточных линий множественной миеломы. Иксазомиб демонстрировал invitro цитотоксичность в отношении клеток миеломы, взятых у пациентов с рецидивом после нескольких предшествующих терапий, включавших бортезомиб, леналидомид и дексаметазон. Комбинация иксазомиба и леналидомида показала синергетическое цитотоксическое действие на клеточные линии множественной миеломы. В условиях invivo иксазомиб проявлял противоопухолевое действие на мышиной модели опухолевого ксенотрансплантата множественной миеломы. В условиях invitro иксазомиб
воздействовал на типы клеток, обнаруженных в микросреде костного мозга, в т.ч. эндотелиальные клетки сосудов, остеокласты и остеобласты.
Кардиоэлектрофизиология
Иксазомиб не удлинял интервал QT в рамках клинически значимого воздействия на основе результатов фармакокинетического и фармакодинамического анализа данных о 245 пациентах. При дозе 4 мг среднее изменение, по сравнению с исходным состоянием интервала QT с корректировкой Фридериция, составило 0,07 мсек (90% ДИ; -0,22, 0,36) согласно анализу, основанному на модели. Не наблюдалось видимой зависимости между концентрацией иксазомиба и интервалом RR, что предполагает отсутствие клинически значимого действия иксазомиба на ритм сердца.
Клиническая эффективность и безопасность
Эффективность и безопасность Нинларо® совместно с леналидомидом и дексаметазоном была проанализирована в ходе международного, рандомизированного, двойного слепого, плацебо контролируемого, мультицентрового исследования по доказательству более высокой эффективности 3 фазы (С16010) у пациентов с рецидивирующей и/или рефрактерной множественной миеломой, которые уже получили не менее одного курса химиотерапии. Всего 722 пациента (популяция больных, включённых в испытание, ITT-популяция) были распределены по группам случайным образом в соотношении 1:1 для проведения курса лечения в виде комбинации Нинларо®, леналидомида и дексаметазона (N = 360; схема лечения с Нинларо®) или плацебо в комбинации с леналидомидом и дексаметазоном (N = 362; схема лечения с плацебо) до прогрессирования заболевания или развития неприемлемой токсичности. У пациентов, включенных в исследование, наблюдалась рефрактерная множественная миелома, в т.ч. первичная рефрактерная; миелома, которая рецидивировала после предшествующей терапии; или рецидивировала и была рефрактерной к любой предшествовавшей терапии. В исследовании могли принять участие пациенты, у которых изменились схемы лечения до прогрессирования заболевания, а также те, у кого наблюдались контролируемые сердечнососудистые заболевания. Из исследования 3 фазы исключались пациенты, которые были невосприимчивыми к леналидомиду или ингибиторам протеасом, и пациенты, которые получили более трех предшествующих курсов терапии. Согласно целям данного испытания рефрактерное заболевание было определено как прогрессирование заболевания при лечении или прогрессирование заболевания в течение 60 дней после приема последней дозы леналидомида или ингибитора протеасом. Так как данные по этим пациентам ограничены, рекомендуется внимательная оценка соотношения пользы и риска перед началом применения схемы лечения с Нинларо®.
Всем пациентам обеих групп лечения была рекомендована тромбопрофилактика согласно инструкции по медицинскому применению леналидомида. Комплексная терапия такими лекарственными средствами как противорвотные, противовирусные и антигистаминные средства назначалась пациентам на усмотрение врача в качестве профилактики и/или лечения симптомов.
Пациенты принимали Нинларо® 4 мг или плацебо в 1, 8 и 15 дни совместно с леналидомидом (25 мг) в дни с 1 по 21 и с дексаметазоном (40 мг) в 1, 8, 15 и 22 дни 28-дневного цикла. Пациенты с почечной недостаточностью получили начальную дозу леналидомида согласно его инструкции по медицинскому применению. Курс лечения продолжался до прогрессирования заболевания или развития неприемлемой токсичности.
В двух исследовательских схемах лечения исходные демографические данные и характеристики заболевания были сбалансированы и сравнимы. Средний возраст составил 66 лет, возрастной диапазон 38-91 год; 58% пациентов были старше 65 лет. Пятьдесят семь процентов пациентов были мужского пола. Восемьдесят пять процентов популяции были представителями белой расы, 9% - азиатской расы и 2% - чернокожими. Общее состояние онкологического больного по шкале ECOG у девяноста трех процентов пациентов составляло 0-1, и согласно Международной системе стадирования (ISS) 12% пациентов в начале исследования имели заболевание 3 степени (N = 90). У двадцати пяти процентов пациентов клиренс креатинина составлял < 60 мл/мин. У двадцати трех процентов пациентов наблюдалась болезнь легких цепей, и у 12% пациентов наблюдалась измеряемая болезнь только при определении свободных легких цепей. У девятнадцати процентов наблюдались цитогенетические аномалии высокого риска (del[17], t[4;14], t[14;16]) (N = 137), 10% имели del(17) (N = 69) и 34% имели амплификацию lq (lq21) (N = 247). Пациенты получили от одного до трех предшествующих курсов терапии (в среднем 1), в т.ч. предшествующий курс лечения бортезомибом (69%), карфилзомибом (< 1%), талидомидом (45%), леналидомидом (12%), мелфаланом (81%). Пятидесяти семи процентам пациентов ранее была проведена трансплантация стволовых клеток. Семьдесят семь процентов пациентов имели рецидив после предшествующего лечения и 11% были рефрактерны к предшествующему лечению. У 6% пациентов наблюдалась первичная резистентность, определяемая как стабилизация заболевания (как лучшая ответная реакция) или прогрессирование заболевания при всех предшествующих курсах терапии.
Первичной конечной точкой была выживаемость без прогрессирования заболевания (ВБПЗ) согласно единым критериям ответа Международной группы по изучению множественной миеломы (IMWG) 2011 года согласно оценке независимого наблюдательного комитета (IRC) на основе результатов центральной лаборатории. Эффективность лечения оценивалась каждые 4 недели до прогрессирования заболевания. При первичном анализе (медиана наблюдения 14,7 месяцев и медиана циклов 13), ВБПЗ статистически значимо отличалась между группами лечения. Обобщенные результаты ВБПЗ приведены в Таблице 4 и на Рис. 1. Улучшения в ВБПЗ при схеме лечения с Нинларо® были подтверждены улучшениями в суммарной эффективности терапии.
Таблица 4. Результаты ВБПЗ и эффективности лечения пациентов с множественной миеломой, получивших курс лечения Нинларо® или плацебо совместно с леналидомидом и дексаметазоном (ITT-популяция)
| Нинларо® + леналидомид и дексаметаззон (N = 360) | Плацебо + леналидомид и дексаметаззон (N = 362) | ||||||
| Выживаемость без прогрессирования заболевания (ВБПЗ) | |||||||
| Случаи ВБПЗ, n (%) | 129 (36) | 157(43) | |||||
| Медиана (месяцы) | 20,6 | 14,7 | |||||
| Значение р* | 0,012 | ||||||
| Соотношение рисков† | 0,74 | ||||||
| (95% ДИ) | (0,59; 0,94) | ||||||
| Общая частота ответа‡, n (%) | 282 (78,3) | 259 (71,5) | |||||
| Категория ответа, n (%) | |||||||
| Полный ответ | 42(11,7) | 24 (6,6) | |||||
| Очень хороший частичный ответ | 131 (36,4) | 117 (32,3) | |||||
| Частичный ответ | 109 (30,3) | 118(32,6) | |||||
| Время до наступления ответа, месяцы | |||||||
| Медиана | 1,1 | 1,9 | |||||
| Продолжительность ответа§, месяцы | |||||||
| Медиана | 20,5 | 15,0 | |||||
* Значение р основано на стратифицированном лог-ранговом критерии.
† Соотношение рисков основано на стратифицированной модели регрессии пропорциональных рисков Кокса. Соотношение рисков меньше чем 1 указывает на превосходство схемы с лекарственным средством Нинларо®.
‡ Общая частота ответа = Полный ответ + Очень хороший частичный ответ + Частичный ответ
§ На основе ответивших на терапию в популяции, подлежащей оценке по восприимчивости к лечению.
Рис. 1. График Каплана-Майера выживаемости без прогрессирования заболевания у ITT- популяции
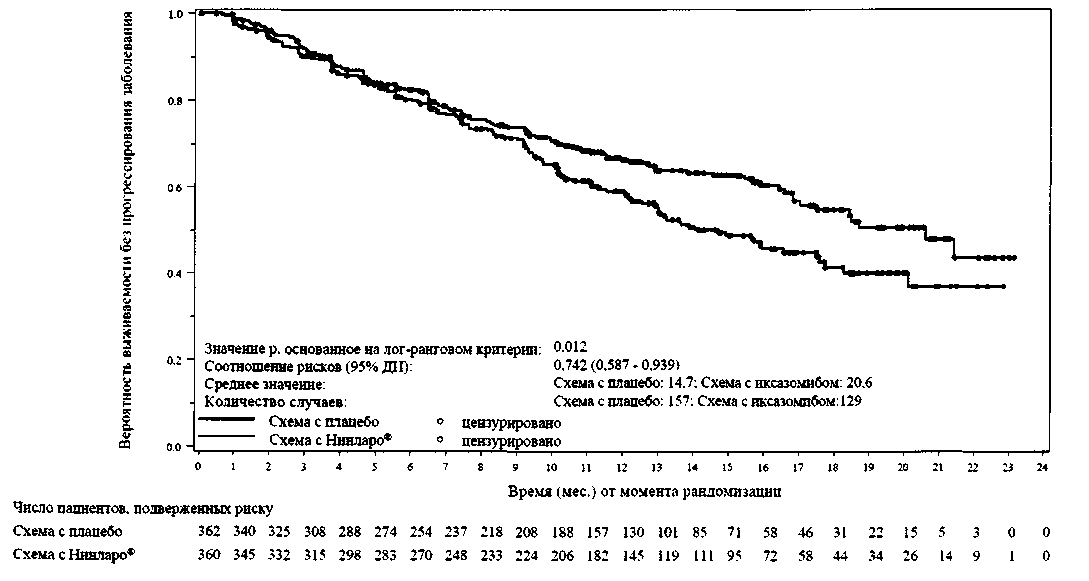
Запланированный промежуточный анализ общей выживаемости (ОВ) в середине периода наблюдения (23 месяца) был проведен с 35% от необходимого количества летальных исходов для окончательного анализа общей выживаемости у 1ТТ-популяции; 81 летальный исход произошел при схеме лечения с Нинларо® и 90 летальных исходов при схеме лечения с плацебо. При обеих схемах лечения медиана общей выживаемости достигнута не была. Согласно проведенному анализу, расчетная медиана ВБПЗ у ITT-популяции составила 20 месяцев при схеме лечения с Нинларо® и 15,9 месяцев при схеме лечения с плацебо (HR=0,82(95% ДИ(0,67; 1,0)]).
Рандомизированное, двойное слепое плацебо-контролируемое исследование 3 фазы было проведено в Китае (N = 115) с аналогичным дизайном исследования и критериями отбора. Многие из пациентов, включенных в исследование, имели при первоначальном диагнозе распространённое заболевание со стадией Durie-Salmon III (69%), получали, по меньшей мере, 2 предшествующие терапии (60%) и были рефрактерными к талидомиду (63%). При первичном анализе (медиана наблюдения 8 месяцев и медиана 6 циклов) медиана ВБПЗ составляла 6,7 месяца при схеме лечения с Нинларо® и 4 месяца при схеме лечения с плацебо (р = 0,035, HR = 0,60). При окончательном анализе ОВ при медиане наблюдения 19,8 месяцев ОВ была лучше у пациентов, получавших лечение с использованием Нинларо®, по сравнению со схемой лечения с использованием плацебо [р = 0,0014, HR=0,42, 95% ДИ: (0,242; 0,726)].
Так как множественная миелома является гетерогенным заболеванием, благоприятный эффект может быть разным в подгруппах данного исследования 3 фазы (С 16010) (см. Рис. 2).
Рис. 2. Форест-диаграмма выживаемости без прогрессирования заболевания в подгруппах
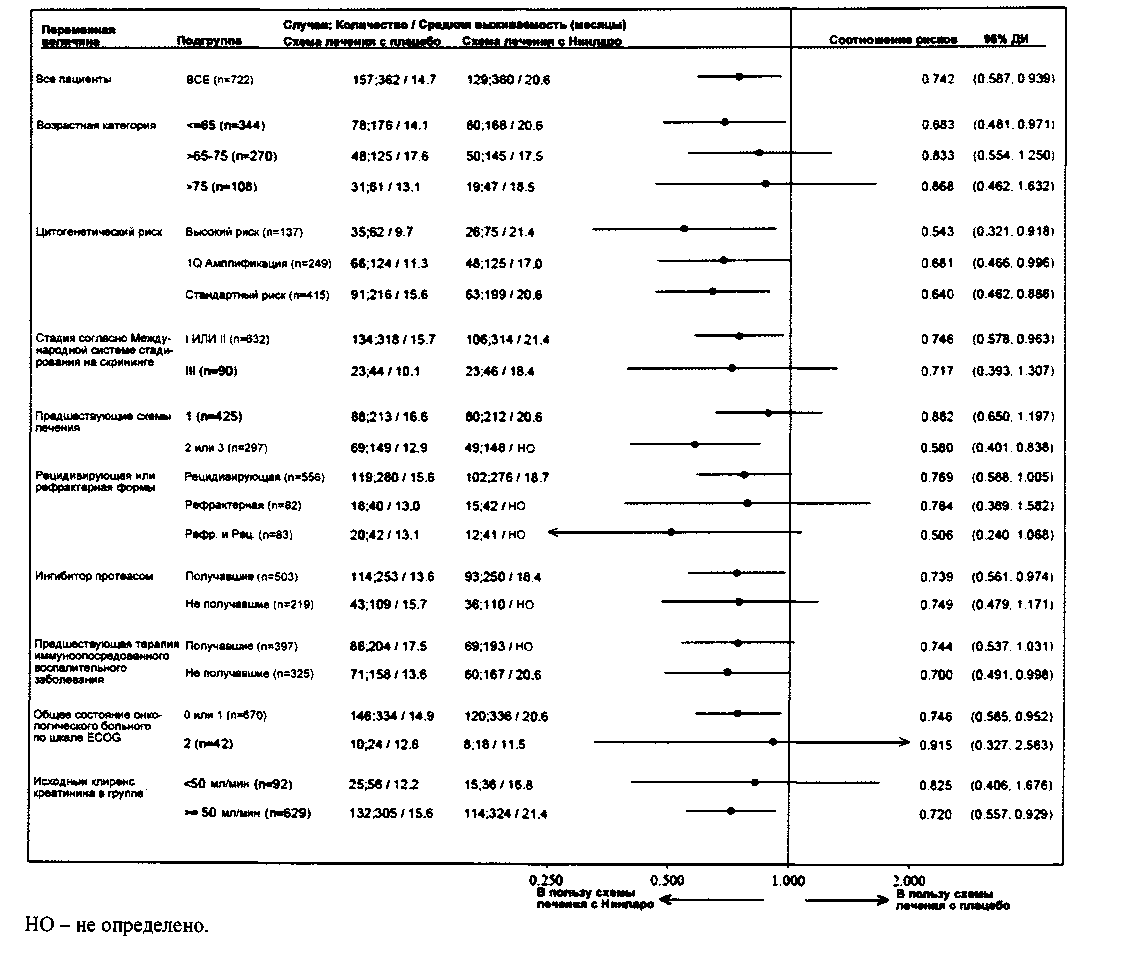
В исследовании 3 фазы (С 16010) у 10 пациентов (по 5 в каждой группе схемы лечения) в начале испытания наблюдалась тяжелая степень почечной недостаточности. Из 5 пациентов при схеме лечения с Нинларо® у одного пациента наблюдался подтвержденный частичный ответ и у 3 пациентов наблюдалась подтвержденная стабилизация заболевания (однако, у 2 из них наблюдался неподтвержденный частичный ответ и у 1 неподтвержденной очень хороший частичный ответ). Из 5 пациентов при схеме лечения с плацебо у 2 наблюдался очень хороший частичный ответ.
Качество жизни, определяемое при помощи методов оценки общего здоровья (опросник QLQ-СЗО и MY-20 Европейской организации по исследованию и лечению онкологических заболеваний (EORTC)), поддерживалось в течение лечения и было сходным в данном исследовании 3 фазы при обеих схемах лечения (С16010).
Дети
Европейское агентство по лекарственным средствам освободило от обязательства предоставлять результаты испытаний Нинларо® во всех подгруппах детской популяции с множественной миеломой (для получения информации о применении в педиатрии см. раздел «Способ применения и дозировка»).
Фармакокинетика
Всасывание
После перорального применения максимальные концентрации иксазомиба в плазме крови достигались приблизительно через час после дозирования. Среднее значение абсолютной биодоступности после перорального приема составляло 58%. AUC иксазомиба увеличивается дозозависимым образом в диапазоне доз от 0,2 до 10,6 мг.
Применение с пищей с высоким содержанием жиров снижало AUC иксазомиба на 28% по сравнению с применением утром натощак (см. раздел «Способ применения и дозировка»).
Распределение
Иксазомиб на 99% связывается с белками плазмы крови и распределяется в эритроцитах в соотношении AUC кровь-плазма, равном 10. Объем распределения в равновесном состоянии составляет 543 л.
Метаболизм
После перорального приема дозы с радиоизотопной меткой иксазомиб представлял 70% всего связанного с лекарственным средством радиоактивного материала в плазме. Основным механизмом выведения иксазомиба считают метаболизм под действием множественных ферментов CYP и не-CYP белков. При клинически значимых уровнях концентрации иксазомиба исследования invitroс использованием изоферментов цитохрома Р450, полученных на основе человеческой комплементарной ДНК, показали, что не существует специфического изофермента CYP, преимущественно участвующего в метаболизме иксазомиба. При концентрациях, более высоких, чем клинические, иксазомиб подвергался метаболизму под действием многих изоформ CYP со следующими оценочными долями участия: 3А4 (42%), 1А2 (26%), 2В6 (16%), 2С8 (6%), 2D6 (5%), 2С19 (5%) и 2С9 (< 1%).
Выведение
Иксазомиб демонстрирует мультиэкспоненциальный фармакокинетический профиль. По результатам популяционного фармакокинетического анализа системный клиренс составлял приблизительно 1,86 л/ч с индивидуальной вариабельностью, составлявшей 44%. Конечный период полувыведения (t1/2) иксазомиба составил 9,5 дней. Приблизительно 2-кратное аккумулирование по AUC наблюдалось при еженедельном пероральном дозировании на 15 день.
Экскреция
После перорального приема однократной дозы 14С-иксазомиба у 5 пациентов с распространённой злокачественной опухолью 62% поступившей радиоактивности было выведено с мочой и 22% с калом. Неизмененный иксазомиб, выведенный с мочой составлял < 3,5% от введенной дозы.
Особые группы пациентов
Печеночная недостаточность
По результатам популяционного фармакокинетического анализа фармакокинетика иксазомиба была сходной у пациентов с нормальной функцией печени и у пациентов с легкой степенью печеночной недостаточности (общий билирубин ≤ ВГН и ACT > ВГН или общий билирубин > 1- 1,5 × ВГН и любое значение ACT).
Фармакокинетика иксазомиба была охарактеризована у пациентов с нормальной функцией печени при дозе 4 мг (N = 12), печеночной недостаточностью средней степени тяжести при дозе
мг (общий билирубин 1,5-3 × ВГН, N = 13) или тяжелой степенью печеночной недостаточностью при дозе 1,5 мг (общий билирубин > 3 × ВГН, N = 18). Значения несвязанной нормализованной по дозе AUC были на 20% выше у пациентов со средней или тяжелой степенью печеночной недостаточности по сравнению с пациентами с нормальной функцией печени (см. раздел «Способ применения и дозировка»).
Почечная недостаточность
По результатам популяционного фармакокинетического анализа фармакокинетика иксазомиба была сходной у пациентов с нормальной функцией почек и у пациентов с легкой или средней степенью почечной недостаточности (клиренс креатинина ≥ 30 мл/мин).
Фармакокинетика иксазомиба была охарактеризована у пациентов с нормальной функцией почек при дозе 3 мг (клиренс креатинина ≥ 90 мл/мин, N = 18), тяжелой степенью почечной недостаточности (клиренс креатинина < 30 мл/мин, N = 14) и при терминальной стадии почечной недостаточности, требующей диализа. Несвязанная AUC была на 38% выше у пациентов с тяжелой степенью почечной недостаточности и при терминальной стадии почечной недостаточности, требующей диализа, по сравнению с пациентами с нормальной функцией почек. Концентрации иксазомиба до и после диализа, измеренные во время сеанса гемодиализа, были схожими, что указывает на то, что иксазомиб не может быть выведен посредством диализа (см. раздел «Способ применения и дозировка»).
Возраст, пол, раса
По результатам популяционного фармакокинетического анализа не наблюдалось клинически значимого влияния возраста (в диапазоне 23-91 года), пола, площади поверхности тела (диапазон 1,2-2,7 м2) или расовой принадлежности на клиренс иксазомиба. Среднее значение AUC было на 35% выше у пациентов азиатского происхождения; однако, наблюдалось частичное совпадение значений AUC иксазомиба у пациентов белой и азиатской расы.
Доклинические данные по безопасности
Мутагенность
У иксазомиба не выявлено мутагенного действия при испытании на обратные мутации у бактерий (тест Эймса) или кластогенного действия при микроядерном испытании на клетках костного мозга у мышей. Иксазомиб показал положительный результат invitro при испытании кластогенности лимфоцитов периферической крови человека. В то же время иксазомиб показал отрицательный результат при кометном анализе invivo у мышей, в котором процент ДНК в хвосте был проанализирован в желудке и печени. Таким образом, весомые доказательства указывают на то, что лекарственное средство Нинларо® не рассматривается как несущее риск генотоксичности.
Репродуктивное развитие и развитие эмбриона и плода
Иксазомиб стал причиной токсичности эмбриона и плода у беременных крыс и крольчих только при дозах, токсичных для материнского организма, и при уровне воздействия, которое было немного выше, чем наблюдаемое у пациентов, получающих рекомендованную дозу. Испытания фертильности и раннего развития эмбриона, пре- и постнатальной токсикологии с иксазомибом не проводились, но был проведен анализ репродуктивных тканей в испытаниях общей токсичности. В результате лечения иксазомибом не наблюдалось влияния на репродуктивные органы особей мужского и женского пола в испытаниях, продолжавшихся до 6 месяцев у крыс и до 9 месяцев у собак.
Токсикология и/или фармакология у животных
В многоцикловых испытаниях токсического действия при многократном введении у крыс и собак главными таргетными органами являлись желудочно-кишечный тракт, лимфоидные ткани и нервная система. В испытании продолжительностью 9 месяцев (10 циклов) у собак, которым вводилось лекарственное средство перорально в режиме дозирования, повторяющем клиническую схему лечения (28-дневный цикл), микроскопическое нейрональное действие в основном было минимальным по сути и наблюдались только при 0,2 мг/кг (4 мг/м2). Большинство полученных данных о таргетных органах продемонстрировало частичное или полное выздоровления после отмены лечения, за исключением нейрональных данных в люмбальном узле заднего корешка и заднего столба.
После перорального введения исследование распределения в тканях у крыс выявило, что головной и спинной мозг были среди тканей с самыми низкими уровнями, из чего можно сделать вывод, что проникновение иксазомиба через гематоэнцефалический барьер является ограниченным. Однако релевантность этих данных для людей неизвестна.
Доклинические фармакологические испытания по безопасности как invitro (ток калиевых каналов hERG), так и invivo (телеметрические данные у собак после одного перорального введения) не продемонстрировали какого-либо действия иксазомиба на сердечно-сосудистую или респираторную функции при AUC более чем в 8 раз выше, чем клиническое значение.
Нинларо Показания к применению
Нинларо® в комбинации с леналидомидом и дексаметазоном применяется для лечения взрослых пациентов с множественной миеломой, получивших, по крайней мере, один курс предшествующей терапии.
Способ применения Нинларо и дозировка
Лечение должно назначаться и проходить под контролем врача, имеющего опыт лечения множественной миеломы.
Дозировка
Рекомендуемая начальная доза Нинларо® составляет 4 мг, принимаемая внутрь один раз в неделю в 1, 8 и 15 дни 28-дневного цикла лечения.
Рекомендуемая начальная доза леналидомида составляет 25 мг внутрь, принимаемая ежедневно с 1 по 21 день 28-дневного цикла лечения.
Рекомендуемая начальная доза дексаметазона составляет 40 мг, принимаемая в 1, 8, 15 и 22 дни 28-дневного цикла лечения.
Схема применения Нинларо® в комбинации с леналидомидом и дексаметазоном
| 28-дневный цикл (4-недельный цикл) | ||||||||
| Неделя 1 | Неделя 2 | Неделя 3 | Неделя 4 | |||||
| 1 день | 2-7 дни | 8 день | 9-14 дни | 15 день | 16-21 дни | 22 день | 23-28 дни | |
| Нинларо® | ✓ | ✓ | ✓ | |||||
| Леналидомид | ✓ | ✓Ежедневно | ✓ | ✓Ежедневно | ✓ | ✓Ежедневно | ||
| Дексаметазон | ✓ | ✓ | ✓ | ✓ | ||||
✓- прием лекарственного средства
Дополнительную информацию по леналидомиду и дексаметазону смотрите в их инструкциях по медицинскому применению.
Перед началом нового цикла лечения:
абсолютное число нейтрофилов должно составлять ≥ 1 000/мм3;
число тромбоцитов должно составлять ≥ 75 000/мм3;
негематологическая токсичность (по оценке врача) должна восстановиться до исходного состояния пациента или до 1 степени тяжести или ниже.
Лечение следует продолжать до начала прогрессирования заболевания или развития неприемлемой токсичности. Лечение Нинларо® в комбинации с леналидомидом и дексаметазоном продолжительностью более 24 циклов должно быть основано на индивидуальной оценке риска и пользы, так как данные о переносимости и токсичности при применении лекарственного средства продолжительностью более 24 циклов ограничены (см. раздел «Фармакодинамика»).
Указания при пропуске дозы
Если прием дозы Нинларо® задержан или пропущен, дозу следует принимать только в том случае, если следующая запланированная доза будет принята более чем через 72 часа. Пропущенную дозу не следует принимать в течение 72 часов перед следующей запланированной дозой. Не следует принимать двойную дозу для восполнения пропущенной дозы.
Если после приема лекарственного средства возникла рвота, пациенту не следует повторять дозу. Пациент должен продолжить прием лекарственного средства во время следующего запланированного приема.
Указания по изменению дозы
Этапы снижения дозы представлены в Таблице 1, указания по изменению дозы представлены в Таблице 2.
Таблица 1. Этапы снижения дозы Нинларо®
| Рекомендуемая начальная доза* | Первое снижение до | Второе снижение до | Отмена |
| 4 мг | 3 мг | 2,3 мг |
* У пациентов со средней или тяжелой степенью печеночной недостаточности с тяжелой степенью почечной недостаточности или при терминальной стадии почечной недостаточности, требующей диализа, рекомендована сниженная доза 3 мг.
Рекомендуется альтернативный подход к изменению дозы для Нинларо® и леналидомида в случае перекрывающихся профилей токсичности при тромбоцитопении, нейтропении и сыпи. В случае перекрывающихся профилей токсичности первым этапом по изменению дозы является отмена/снижение дозы леналидомида. Следует обратиться к инструкции по медицинскому применению леналидомида, если требуется снижение его дозы.
Таблица 2. Указания по изменению дозы Нинларо® при применении в комбинации с леналидомидом и дексаметазоном
| Гематологическая токсичность | Рекомендуемые действия |
| Тромбоцитопения (количество тромбоцитов) | |
| Количество тромбоцитов менее чем30 000/мм3 | Не применять Нинларо® и леналидомид, пока количество тромбоцитов не станет, по крайней мере, 30 000/мм3.После нормализации продолжить применение леналидомида в следующей более низкой дозе согласно его инструкции по применению и Нинларо® в последней дозе.Если количество тромбоцитов снова падает ниже, чем 30 000/мм3, не применять Нинларо® и леналидомид, пока количество тромбоцитов не станет, по крайней мере, 30 000/мм3.После нормализации продолжить применение Нинларо® в следующей более низкой дозе и леналидомида в последней дозе.* |
| Нейтропения (абсолютное количество нейтрофилов) | |
| Абсолютное количество нейтрофилов менее чем 500/мм3 | Не применять Нинларо® и леналидомид, пока абсолютное количество нейтрофилов не станет, по крайней мере, 500/мм3. Рассмотреть целесообразность применения ГКСФ (гранулоцитарного колониестимулирующего фактора) согласно клиническим рекомендациям.После нормализации продолжить применение леналидомида в следующей более низкой дозе согласно его инструкции по применению и Нинларо® в последней дозе.Если абсолютное количество нейтрофилов снова падает ниже, чем 500/мм3, не применять Нинларо® и леналидомид, пока абсолютное количество нейтрофилов не станет, по крайней мере, 500/мм3.После нормализации продолжить применение Нинларо® в следующей более низкой дозе и леналидомида в его последней дозе * |
| Негематологическая токсичность | Рекомендуемые действия |
| Сыпь | |
| Степень 2 или 3† | Не применять леналидомид, пока сыпь не уменьшится до 1 степени тяжести или ниже.После нормализации продолжить применение леналидомида в следующей более низкой дозе согласно его инструкции по применению.Если появляется сыпь 2 или 3 степени тяжести, не применять Нинларо® и леналидомид, пока сыпь не уменьшится до 1 степени тяжести или ниже.После нормализации продолжить применение Нинларо® в следующей более низкой дозе и леналидомида в его последней дозе. |
| Степень 4 | Отменить схему лечения. |
| Периферическая нейропатия | |
| Периферическая нейропатия1 степени с болевым синдромом или периферическая нейропатия2 степени | Не применять Нинларо®, пока периферическая нейропатия не уменьшится до 1 степени тяжести или ниже без болевого синдрома или до исходного состояния пациента.После нормализации продолжить применение Нинларо® в последней дозе. |
| Периферическая нейропатия2 степени с болевым синдромом или периферическая нейропатия3 степени | Не применять Нинларо®. Реакции токсичности должны получить обратное развитие до исходного состояния пациента или до 1 степени тяжести или ниже (по оценке врача) перед возобновлением приема Нинларо®.После нормализации продолжить применение Нинларо® в следующей более низкой дозе. |
| Периферическая нейропатия4 степени | Отменить схему лечения. |
| Другие виды негематологической токсичности | |
| Другие виды негематологической токсичности 3 или 4 степени | Не применять Нинларо®. Реакции токсичности должны получить обратное развитие до исходного состояния пациента или до 1 степени тяжести или ниже (по оценке врача), перед возобновлением приема Нинларо®.Если реакция связана с приемом Нинларо®, после нормализации продолжить лечение Нинларо® в следующей более низкой дозе. |
*При последующих реакциях чередуют изменение дозы леналидомида и лекарственного средства Нинларо®.
†Оценка степени основана на версии 4.03 Общих терминологических критериев оценки нежелательных реакций (СТСАЕ) Национального института рака США.
Сопутствующее лечение
Следует предусмотреть антивирусную профилактику у пациентов, получающих лечение лекарственным средством Нинларо®, для уменьшения риска повторного развития опоясывающего лишая. Пациенты, участвовавшие в исследованиях с Нинларо®, которые получали антивирусную профилактику, имели более низкий уровень заболевания опоясывающим лишаем, чем те, кто ее не получал.
Профилактика тромбоза рекомендована пациентам, получающим лечение лекарственным средством Нинларо® совместно с леналидомидом и дексаметазоном, и она должна быть основана на оценке основных рисков и клинического состояния пациента.
В отношении иного сопутствующего лечения, которое может потребоваться, необходимо обратиться к инструкциям по медицинскому применению леналидомида и дексаметазона.
Особые группы пациентов
Пожилые люди
Необходимость в коррекции дозы Нинларо® у пациентов старше 65 лет отсутствует.
Были получены сообщения об отмене лечения у пациентов > 75 лет: у 13 пациентов (28%), получавших Нинларо®, и у 10 пациентов (16%), получавших плацебо. У пациентов > 75 лет наблюдалась сердечная аритмия: 10 пациентов (21%), получавших Нинларо®, и 9 пациентов (15%), получавших плацебо.
Печеночная недостаточность
Отсутствует необходимость в коррекции дозы Нинларо® у пациентов с легкой степенью печеночной недостаточности (общий билирубин ≤ верхней границы нормы (ВГН) и аспартатаминотрансфераза (ACT) > ВГН или общий билирубин > 1-1,5 × ВГН и любой уровень ACT). Сниженная доза 3 мг рекомендуется пациентам со средней (общий билирубин > 1,5-3 × ВГН) или тяжелой (общий билирубин > 3 × ВГН) степенью печеночной недостаточности (см. раздел 5.2).
Почечная недостаточность
Отсутствует необходимость в коррекции дозы Нинларо® у пациентов с легкой или средней степенью почечной недостаточности (клиренс креатинина ≥ 30 мл/мин). Сниженная доза 3 мг рекомендуется пациентам с тяжелой степенью почечной недостаточности (клиренс креатинина < 30 мл/мин) и при терминальной стадии почечной недостаточности, требующей диализа. Нинларо® не подвергается диализу и поэтому может применяться независимо от времени его проведения (см. раздел «Фармакокинетика»).
Относительно рекомендаций по применению у пациентов с почечной недостаточностью необходимо обратиться к инструкции по медицинскому применению леналидомида.
Способ применения
Нинларо® предназначено для перорального применения.
Нинларо® необходимо принимать приблизительно в одно и то же время в 1, 8 и 15 дни лечебного цикла за 1 час до или через 2 часа после еды (см. раздел 5.2). Капсулу необходимо проглатывать целиком, запивая водой. Капсулу нельзя ломать, жевать или открывать (см. раздел «Особые указания по применению и меры предосторожности при утилизации»).
Побочные действия
Так как Нинларо® применяется в комбинации с леналидомидом и дексаметазоном, для получения дополнительной информации о нежелательных реакциях необходимо обратиться к их инструкциям по медицинскому применению.
Обзор профиля безопасности
Ниже представлены сводные данные по безопасности, полученные в рамках базового исследования 3 фазы С16010 (N = 720) и двойного слепого плацебо-контролируемого Китайского продолженного исследования С16010 (N = 115). Наиболее часто наблюдаемыми нежелательными реакциями (≥ 20%) среди 417 пациентов, которые получали схему лечения, включающую лекарственное средство Нинларо®, и среди 418 пациентов, которые получали схему лечения с плацебо, были диарея (39% против 32%), запор (30% против 22%), тромбоцитопения (33% против 21%), нейтропения (33% против 30%), периферическая нейропатия (25% против 20%), тошнота (23% против 18%), периферический отек (23% против 17%), рвота (20% против 10%) и инфекции верхних дыхательных путей (21% против 16%). Сообщалось о развитии серьезных нежелательных реакций у ≥ 2% пациентов, включающих тромбоцитопению (2%) и диарею (2%).
Табулированный перечень нежелательных реакций
Классификация частоты нежелательных реакций (HP) определяется следующим образом: очень часто (≥ 1/10); часто (≥ 1/100 до < 1/10); нечасто (≥ 1/1 000 до < 1/100); редко (≥ 1/10 000 до < 1/1 000); очень редко (< 1/10 000); частота неизвестна (невозможно оценить на основании полученных данных). В каждом системно-органном классе нежелательные реакции сгруппированы по частоте возникновения, самые частые реакции указываются первыми. Внутри каждой частотной группы нежелательные реакции приведены в порядке убывания серьезности.
Таблица 3. Нежелательные реакции, наблюдавшиеся у пациентов, принимающих Нинларо® совместно с леналидомидом и дексаметазоном (все степени тяжести, 3 и 4 степени тяжести)
| Системно-органный класс/нежелательная реакция | Нежелательные реакции (все степени тяжести) | Нежелательные реакции 3 степени тяжести | Нежелательные реакции 4 степени тяжести |
| Инфекционные и паразитарные заболевания | |||
| Инфекция верхних дыхательных путей | Очень часто | Нечасто | |
| Опоясывающий лишай | Часто | Часто | |
| Нарушения со стороны крови и лимфатической системы | |||
| Тромбоцитопения * | Очень часто | Очень часто | Часто |
| Нейтропения | Очень часто | Очень часто | Часто |
| Нарушение со стороны нервной системы | |||
| Периферические нейропатии* | Очень часто | Часто | |
| Нарушение со стороны желудочно-кишечного тракта | |||
| Диарея | Очень часто | Часто | |
| Тошнота | Очень часто | Часто | |
| Рвота | Очень часто | Нечасто | |
| Запор | Очень часто | Нечасто | |
| Нарушения со стороны кожи и подкожных тканей | |||
| Сыпь* | Очень часто | Часто | |
| Нарушения со стороны мышечной, скелетной и соединительной ткани | |||
| Боль в спине | Очень часто | Нечасто | |
| Общие нарушения и реакции в месте введения | |||
| Периферический отек | Очень часто | Часто | |
Примечание: нежелательные реакции, включенные в виде терминов предпочтительного употребления, основаны на версии 16.0 MedDRA (Медицинский словарь терминов регламентирующей деятельности).
* Представляет собой объединение терминов предпочтительного употребления.
Описание отдельных нежелательных реакций
Прекращение лечения
У ≤ 1% пациентов, получавших схему лечения с Нинларо®, в связи с развитием нежелательных реакций отменяли одно или более из трех применяемых лекарственных средств.
Тромбоцитопения
У 3% пациентов, получавших схему лечения с Нинларо®, и у 1% пациентов, получавших схему лечения с плацебо, во время лечения отмечалось количество тромбоцитов ≤ 10 000/мм3. Менее чем у 1% пациентов на фоне использования обеих схем лечения отмечалось количество тромбоцитов ≤ 5 000/мм3. У < 1% пациентов, получавших схему лечения с Нинларо®, и у 2% пациентов, получавших схему лечения с плацебо, отменяли одно или более из трех применяемых лекарственных средств вследствие развития тромбоцитопении. Развитие тромбоцитопении не привело к увеличению случаев геморрагических событий или увеличению частоты переливания тромбоцитарной массы.
Желудочно-кишечная токсичность
Диарея привела к отмене одного или более из трех применяемых лекарственных средств у 1% пациентов при применении схемы лечения с Нинларо®, и у < 1% пациентов при применении схемы лечения с плацебо.
Сыпь
Сыпь развилась у 18% пациентов, получавших схему лечения с Нинларо®, по сравнению с 10% пациентов, получавших схему лечения с плацебо. Наиболее частым видом сыпи при применении обеих схем являлась макулопапулезная и макулезная сыпь. Сыпь 3 степени тяжести отмечена у 2% пациентов при применении схемы лечения с Нинларо® по сравнению с 1% пациентов при применении схемы лечения с плацебо. Сыпь приводила к отмене одного или более из трех применяемых лекарственных средств у < 1% пациентов при обеих схемах.
Периферическая нейропатия
Периферическая нейропатия развилась у 25% пациентов при применении схемы лечения с Нинларо®, по сравнению с 20% пациентов при схеме лечения с плацебо. Нежелательные реакции в виде периферической нейропатии 3 степени тяжести развились у 2% пациентов в обеих схемах лечения. Наиболее частой нежелательной реакцией была периферическая сенсорная нейропатия (16% и 12% при применении схем с Нинларо® и плацебо, соответственно). Развитие периферической моторной нейропатии наблюдалось в обеих схемах нечасто (< 1%). Периферическая нейропатия привела к отмене одного или более из трех применяемых лекарственных средств у 1% пациентов при применении схемы лечения с лекарственным средством Нинларо® и у < 1% пациентов при применении схемы лечения с плацебо.
Нарушения со стороны органа зрения
Нарушения со стороны органа зрения описывали различными терминами предпочтительного употребления, в общей сложности их частота составляла 24% у пациентов, получавших схему лечения с Нинларо®, и 15% у пациентов, получавших схему лечения с плацебо. Наиболее частыми нежелательными реакциями была нечеткость зрения (5% при схеме лечения с Нинларо®, и 4% при схеме лечения с плацебо), сухость глаз (4% при схеме лечения с Нинларо® и 1% при схеме лечения с плацебо), конъюнктивит (5% при схеме лечения с Нинларо®, и 1% при схеме лечения с плацебо) и катаракта (4% при схеме лечения с Нинларо®, и 5% при схеме лечения с плацебо). Нежелательные реакции 3 степени тяжести наблюдались у 2% пациентов при обеих схемах.
Другие нежелательные реакции
Вне 3 фазы исследования были получены сообщения о следующих серьезных редко встречающихся нежелательных реакциях: острый фебрильный нейтрофильный дерматоз (синдром Свита), синдром Стивенса-Джонсона, поперечный миелит, синдром задней обратимой энцефалопатии, синдром лизиса опухоли и тромботическая тромбоцитопеническая пурпура. Согласно сводным данным по безопасности, полученным в рамках базового исследования 3 фазы С16010 (N = 720) и двойного слепого плацебо-контролируемого Китайского
продолженного исследования С16010 (N = 115) следующие нежелательные реакции были зарегистрированы со схожей частотой встречаемости при схеме лечения с Нинларо® и при схеме лечения с плацебо: усталость (26% против 24%), снижение аппетита (12% против 9%), гипотензия (4% в обеих группах), сердечная недостаточность* (3% в обеих группах), аритмия* (12% в против 11%) и печеночная недостаточность, включая изменение активности ферментов* (8% против 6%).
Частота встречаемости гипокалиемии тяжелой степени (3-4 степени) была выше у пациентов при схеме лечения с Нинларо® (5%), чем при схеме лечения с плацебо (≤ 1%).
У пациентов, получавших Нинларо® совместно с леналидомидом и дексаметазоном, были зарегистрированы редкие случаи развития грибковой и вирусной пневмонии, которые привели к летальному исходу.
Противопоказания Нинларо
Гиперчувствительность к активному веществу или к любому из вспомогательных веществ, перечисленных в пункте «Перечень вспомогательных веществ».
Так как Нинларо® применяется совместно с леналидомидом и дексаметазоном, для получения дополнительной информации о противопоказаниях необходимо обратиться к их инструкциям по медицинскому применению.
Передозировка
Специфического антидота при передозировке лекарственным средством Нинларо® не существует. Клинические данные о передозировке ограничены, однако в рамках рандомизированного контролируемого исследования были получены сообщения о максимальных однократных дозах до 12 мг. В случае передозировки необходимо следить за появлением нежелательных реакций (раздел «Побочное действие») и осуществлять соответствующую поддерживающую терапию.
Меры предосторожности
Так как Нинларо® назначается совместно с леналидомидом и дексаметазоном, для получения дополнительной информации о мерах предосторожности необходимо обратиться к их инструкциям по медицинскому применению.
Тромбоцитопения
Имеются сообщения о развитии тромбоцитопении при применении Нинларо®, с минимальным числом тромбоцитов, как правило, между 14-21 днями каждого 28-дневного цикла, и восстановлением до исходного уровня к началу следующего цикла терапии (см. раздел «Побочное действие»).
Во время лечения Нинларо® количество тромбоцитов следует контролировать не реже одного раза в месяц. Рекомендуется рассмотреть более частый контроль в течение первых трех циклов терапии в соответствии с инструкцией по медицинскому применению леналидомида.
Тромбоцитопения корригируется путем модификации дозы (см. раздел «Способ применения и дозировка») и переливанием тромбоцитарной массы согласно стандартным клиническим рекомендациям.
Желудочно-кишечная токсичность
При применении Нинларо® сообщалось о развитии диареи, запора, тошноты и рвоты, которые в редких случаях требовали применения противодиарейных и противорвотных средств, а также поддерживающей терапии (см. раздел «Побочное действие»). Необходимо корректировать дозу лекарственного средства при развитии симптомов 3 или 4 степени тяжести (см. раздел «Способ применения и дозировка»). В случае развития тяжелых желудочно-кишечных реакций рекомендуется контролировать уровень сывороточного калия.
Периферическая нейропатия
Имеются сообщения о развитии периферической нейропатии при применении Нинларо® (см. раздел «Побочное действие»). Следует проверять пациентов на наличие симптомов периферической нейропатии. Пациентам, у которых появились симптомы периферической нейропатии или ухудшилось течение уже имеющейся периферической нейропатии, может потребоваться модификация дозы (см. раздел «Способ применения и дозировка»).
Периферический отек
Сообщалось о развитии периферических отеков при применении Нинларо® (см. раздел «Побочное действие»). Следует оценить первопричины и назначить, при необходимости, поддерживающее лечение. При развитии симптомов 3 или 4 степени тяжести корректируют дозу дексаметазона согласно его инструкции по медицинскому применению или дозу Нинларо® (см. раздел «Способ применения и дозировка»).
Кожные реакции
Сообщалось о развитии сыпи при применении Нинларо® (см. раздел «Побочное действие»). В случае развития сыпи 2 степени тяжести или выше данная реакция купируется путем назначения поддерживающей терапии или модификации дозы (см. раздел «Способ применения и дозировка»).
Гепатотоксичность
Сообщалось о редких случаях развития лекарственного поражения печени, гепатоцеллюлярного повреждения, жировой дистрофии печени, холестатического гепатита и гепатотоксичности у пациентов, получавших Нинларо® (см. раздел «Побочное действие»). Следует регулярно контролировать уровень печеночных ферментов и корригировать дозу при развитии симптомов 3 или 4 степени тяжести (см. раздел «Способ применения и дозировка»).
Беременность
Женщинам детородного возраста следует избегать наступления беременности в период применения Нинларо®. Если лекарственное средство Нинларо® применяется в период беременности или беременность наступает во время его применения, пациентку следует проинформировать о потенциальной опасности для плода.
Женщинам детородного возраста следует использовать методы эффективной контрацепции в период лечения Нинларо® и в течение 90 дней после приема последней дозы (см. разделы «Взаимодействие с другими лекарственными средствами» и «Период беременности и грудного вскармливания»). Женщины, принимающие гормональные контрацептивы, должны дополнительно применять барьерный метод контрацепции.
Синдром задней обратимой энцефалопатии
Имеются сообщения о развитии синдрома задней обратимой энцефалопатии (ЗОЭ) при применении Нинларо®. Синдром ЗОЭ является редким обратимым неврологическим нарушением, при котором могут наблюдаться судороги, гипертензия, головная боль, нарушение сознания и нарушение зрения. Для подтверждения диагноза проводится томография головного мозга, предпочтительно МРТ. Необходимо отменить схему лечения с Нинларо® при развитии синдрома ЗОЭ.
Мощные индукторы CYP3A
Мощные индукторы могут снизить эффективность Нинларо®, по этой причине следует избегать совместного применения таких сильных индукторов CYP3A, как карбамазепин, фенитоин, рифампицин и зверобой (Hypericumperforatum) (см. разделы «Взаимодействие с другими лекарственными средствами» и «Фармакокинетика»). При отсутствии альтернативы комбинированного применения с мощными индукторами CYP3A, следует осуществлять тщательный мониторинг эффективности контроля заболевания.
Дети
Безопасность и эффективность применения Нинларо® у детей младше 18 лет не установлена. Данные отсутствуют.
Применение в период беременности и кормления грудью
Так как Нинларо® применяется в комбинации с леналидомидом и дексаметазоном, для получения дополнительной информации по фертильности, беременности и грудному вскармливанию необходимо обратиться к их инструкциям по медицинскому применению.
Женщины детородного возраста/Контрацепция у мужчин и женщин
Пациенты мужского и женского пола, которые могут иметь детей, должны использовать высокоэффективные методы контрацепции в период применения Нинларо® и в течение 90 дней после завершения лечения. Нинларо® не рекомендован женщинам детородного возраста, не использующим контрацепцию.
При применении Нинларо® совместно с дексаметазоном, который известен как слабый или средний индуктор CYP3A4, также как и других ферментов и транспортеров, необходимо учитывать риск снижения эффективности оральных контрацептивов. Женщины, принимающие гормональные контрацептивы, должны дополнительно применять барьерный метод контрацепции.
Беременность
Лекарственное средство Нинларо® не рекомендовано во время беременности, так как оно может оказать вредное воздействие на плод при применении у беременных женщин. По этой причине женщины должны избегать наступления беременности в период лечения Нинларо®.
Данные о применении Нинларо® у беременных женщин отсутствуют. Испытания на животных продемонстрировали репродуктивную токсичность (см. раздел «Доклинические данные по безопасности»).
Нинларо® применяют совместно с леналидомидом, леналидомид структурно связан с талидомидом. Талидомид является известным тератогенным активным веществом, которое вызывает тяжелые угрожающие жизни врожденные дефекты у человека. Если леналидомид принимается во время беременности, он оказывает ожидаемое тератогенное действие на организм человека. Условия программы по предупреждению беременности для леналидомида должны выполняться всеми пациентами, за исключением случаев, когда есть надежное доказательство того, что у пациента отсутствует репродуктивный потенциал. Пожалуйста, обратитесь к инструкции по медицинскому применению леналидомида.
Грудное вскармливание
Неизвестно, проникает ли Нинларо® и его метаболиты в грудное молоко. Данные об испытаниях на животных отсутствуют. Рекомендуется прекратить грудное вскармливание, так как риск для новорожденных/младенцев не может быть исключен.
Так как Нинларо® принимается совместно с леналидомидом, грудное вскармливание должно быть прекращено из-за применения леналидомида.
Фертильность
Испытания фертильности с Нинларо® не проводились (см. раздел «Доклинические данные по безопасности»).
Способность влиять на скорость реакции при управлении транспортом
Нинларо® оказывает незначительное влияние на способность управлять транспортными средствами и другими механизмами. В клинических исследованиях наблюдалась усталость и головокружение. Если пациенты испытывают какие-либо из этих симптомов, управлять транспортными средствами или другими механизмами не рекомендуется.
Взаимодействие с другими лекарственными средствами
Фармакокинетическое взаимодействие
Ингибиторы CYP
Комплексное применение иксазомиба с кларитромицином, сильным ингибитором цитохрома CYP3A, не приводит к клинически значимому изменению в системном воздействии иксазомиба. Показатель Сmах иксазомиба снизился на 4% и AUC (площадь под фармакокинетической кривой зависимости концентрации от времени) увеличилась на 11%. Таким образом, коррекция дозы не требуется при комплексной терапии иксазомибом с сильными ингибиторами CYP3A.
На основе результатов анализа фармакокинетики в популяции комплексное применение иксазомиба с сильными ингибиторами CYP1A2 не привело к клинически значимому изменению в системном воздействии иксазомиба. Таким образом, коррекция дозы иксазомиба не требуется при совместном назначении с сильными ингибиторами CYP1А2.
Индукторы CYP
Комплексная терапия иксазомиба с рифампицином снизила Сmах иксазомиба на 54% и AUC на 74%. Таким образом, комплексная терапия с применением сильных индукторов CYP3A с иксазомибом не рекомендуется (см. раздел «Меры предосторожности»).
Влияние иксазомиба на другие лекарственные средства
Иксазомиб не является обратимым или зависящим от времени ингибитором CYP: 1А2, 2В6, 2С8, 2С9, 2С19, 2D6 или 3А4/5. Иксазомиб не индуцировал активность CYP1А2, CYP2В6 и CYP3A4/5 или соответствующие иммунореактивные белковые уровни. Не ожидается, что иксазомиб будет вызывать лекарственные взаимодействия через ингибирование или индуцирование CYP.
Взаимодействия на основе транспортера
Иксазомиб является низкоаффинным субстратом гликопротеина-Р. Иксазомиб не является субстратом белка резистентности рака молочной железы (BCRP), белка, ассоциированного с множественной лекарственной резистентностью 2 (MRP2), или печеночных транспортных полипептидов органических анионов (ОАТР). Иксазомиб не является ингибитором гликопротеина-Р, BCRP, MRP2, ОАТР1В1, ОАТР1В3, ОСТ2, ОАТ1, ОАТ3, МАТЕ1 или МАТЕ2-К. Не ожидается, что иксазомиб будет вызывать лекарственные взаимодействия, опосредованные транспортером.
Оральные контрацептивы
При применении Нинларо® совместно с дексаметазоном, который известен как слабый или средний индуктор CYP3A4, также как и других ферментов и транспортеров, необходимо учитывать риск снижения эффективности оральных контрацептивов. Женщины, принимающие гормональные контрацептивы, должны дополнительно применять барьерный метод контрацепции.
Условия и срок хранения Нинларо
3 года.
Не использовать по истечению срока годности, указанного на упаковке.
Хранить при температуре не выше 30°С. Не замораживать.
Хранить в оригинальной упаковке непосредственно до приема каждой капсулы.
Хранить в недоступном для детей месте.
Упаковка
По 1 капсуле в ПВХ Алюминий/Алюминий блистер, вклеенный в складывающуюся картонную обложку (блистер герметично запаян в картонную обложку и является ее неотделимой частью). По 1 картонной обложке вместе с инструкцией по применению помещают в пачку картонную (промежуточную упаковку). 3 картонные пачки (промежуточные упаковки) помещают в общую картонную пачку. На защитной наклейке голографическим способом указывают логотип «Takeda».
и/или
По 1 капсуле в ПВХ Алюминий/Алюминий блистер, вклеенный в складывающуюся картонную обложку (блистер герметично запаян в картонную обложку и является ее неотделимой частью). По 1 картонной обложке вместе с инструкцией по применению помещают в пачку картонную.
Особые указания по применению и меры предосторожности при утилизации
Лекарственное средство Нинларо® является цитотоксическим. Капсулу необходимо вынимать из упаковки непосредственно перед приемом. Не следует открывать или разрушать капсулы. Необходимо избегать непосредственного контакта с содержимым капсулы. В случае вскрытия капсулы необходимо избегать образования пыли в воздухе во время уборки. При контакте тщательно смыть водой с мылом.
Любое неиспользованное количество лекарственного средства или его отходов следует уничтожать в соответствии с местными требованиями.
Правила отпуска
По рецепту врача.
Информация о производителе
Такеда Фарма А/С, Дания.
Дюбендаль Алле 10,2630 Тоструп.
Takeda Pharma A/S, Denmark.
Dubendal Alle 10, 2630 Taastrup.
Претензии потребителей на территории Республики Беларусь направлять по адресу
Представительство ООО «Takeda Osteuropa Holding GmbH» (Австрийская Республика) в Республике Беларусь.
пр-т Победителей, 84, офис 27, 220020, Минск, Республика Беларусь.
тел. +375 17 240 41 20, факс +375 17 240 41 30.
Информацию о нежелательных реакциях направлять по адресу
Республиканское унитарное предприятие «Центр экспертиз и испытаний в здравоохранении».
Товарищеский пер., 2а, 220037, Минск, Республика Беларусь.
Дальнейшая информация
Помните, храните эти и все другие лекарства в недоступном для детей месте, никогда не передавайте свои лекарства другим и используйте Нинларо только по назначению врача.
Всегда консультируйтесь со своим врачом, чтобы убедиться, что информация, которая отображается на этой странице, может быть применена к вашим личным обстоятельствам.
Внимание! Эта инструкция по применению лекарственного средства является официальной инструкцией производителя Haupt Pharma Amareg GmbH, Германия/ AndersonBrecon (UK) Limited, Великобритания/ Eurofins Biopharma Product Testing Ireland Limited, Ирландия/ Takeda Ireland Ltd..
Авторское право:
- Haupt Pharma Amareg GmbH, Германия/ AndersonBrecon (UK) Limited, Великобритания/ Eurofins Biopharma Product Testing Ireland Limited, Ирландия/ Takeda Ireland Ltd.
| Тип данных | Сведения из реестра |
| Торговое наименование: | Нинларо |
| Форма выпуска: | капсулы 2,3мг, 3мг, 4мг в блистерах в упаковке №1х1, №1х3 |
| Международное наименование: | Ixazomib |
| Производитель: | Haupt Pharma Amareg GmbH, Германия/ AndersonBrecon (UK) Limited, Великобритания/ Eurofins Biopharma Product Testing Ireland Limited, Ирландия/ Takeda Ireland Ltd., Ирландия |
| Заявитель: | Millennium Pharmaceuticals, Inc., США |
| Номер регистрации: | 10574/17/19/20 |
| Дата регистрации: | 28.06.2017 |
| Срок действия: | 28.06.2022 |
| Дата переоформления: | 07.02.2020 |
| Тип: | Лекарственное средство |
| Оригинальное: | оригинальное |
| Состав лекарственного средства: | Ixazomib |
| Код АТХ: | L01XX50 |
| Производитель готовой лекарственной формы: | Haupt Pharma Amareg GmbH, Германия |
| Производитель, осуществляющий фасовку/упаковку: | AndersonBrecon (UK) Limited, Великобритания |
| Контроль качества: | |
| Выдача разрешения на выпуск лекарственного средства: | Takeda Ireland Ltd., Ирландия |
| Другие участники производства: | Eurofins Biopharma Product Testing Ireland Limited, Ирландия - проведение испытаний при выпуске |
| Заявленная цена: | №1 - 1660; №3 - 4980EUR |
| Порядок отпуска: | по рецепту |
| Список хранения: | |
| Срок годности лекарства: | 3 года |
| Нормативная документация: | НД РБ 9174-2017 |
| Дата утверждения нормативной документации: | 28 июня 2017 г. 0:00 |
| Срок действия нормативной документации: | 28 июня 2022 г. 0:00 |
| Изменение в нормативной документации: | Изменение по разделу "Производитель" (изменение производителя, осуществляющего выпуск серий ГЛС) с согласованием дизайна первичных и вторичных упаковок (пр. №691 от 02.07.2020) изменение наименования производителя, осуществляющего проведение испытаний при выпуске лекарственного средства (пр. №1196 от 08.10.2019) Изменение по разделам "НД по контролю качества готового лекарственного средства " (показатель "Вода") и "НД по контролю качества активной фармацевтической субстанции" (пр. №805 от 27.06.2019) Изменение по разделу "Вода" для готового лекарственного средства и по разделам "Описание", "Количественное определение и примеси методом ВЭЖХ", "Энатиомерная примесь методом хиральной ВЭЖХ", "Подлинность и содержание иксазомиба методом ВЭЖХ", "Подлинность методом инфракрасной спектроскопии с Фурье-преобразованием", "Размер части", "Полиморфная форма", "Содержание воды" для активной фармацевтической субстанции (пр. №805 от 27.06.2019) Изменение по разделам "НД по контролю качества активной субстанции", "Производитель активной субстанции" (пр. №343 от 11.04.2018) |
| Номер разрешения НД: | 11213 |
| Код АТХ | Название группы |
| L | Противоопухолевые препараты и иммуномодуляторы |
| L01 | Противоопухолевые препараты |
| L01X | Другие противоопухолевые препараты |
| L01XX | Прочие противоопухолевые препараты |
| L01XX50 | Ixazomib |