

- Склад
- Лікарська форма
- Фармакотерапевтична група
- Фармакологічні властивості
- Клінічні характеристики
- Показання
- Протипоказання
- Взаємодія з іншими засобами
- Особливості застосування
- У період вагітності
- При керуванні автомобілем
- Спосіб застосування та дози
- Діти
- Передозування
- Побічні реакції
- Термін придатності
- Умови зберігання
- Упаковка
- Категорія відпуску
- Виробник
- Місцезнаходження виробника
Луцентіс інструкція із застосування
Офіційна інструкція лікарського засобу Луцентіс розчин 0,23 мл. Опис та застосування Lutsentіs, аналоги та відгуки. Інструкція Луцентіс розчин затверджена виробником.
Склад
діюча речовина: ranibizumab;
1 мл розчину містить 10 мг ранібізумабу;
допоміжні речовини: α,α-трегалози дигідрат; L-гістидину гідрохлорид, моногідрат; L-гістидин; полісорбат 20; вода для ін’єкцій.
Лікарська форма
Розчин для ін’єкцій.
Основні фізико-хімічні властивості: прозорий або ледь опалесціюючий розчин.
Фармакотерапевтична група
Засоби, що застосовуються при судинних захворюваннях очей. Антинеоваскуляризаційні засоби.
Код АТХ S01L A04.
Фармакологічні властивості
Фармакодинаміка.
Ранібізумаб — фрагмент рекомбінантного гуманізованого моноклонального антитіла проти людського судинного ендотеліального фактора росту А (VEGF-A). Він має високу спорідненість з ізоформами VEGF-A (наприклад VEGF110, VEGF121 та VEGF165) і, таким чином, запобігає прикріпленню VEGF-A до його рецепторів VEGFR-1 та VEGFR-2. Прикріплення VEGF-A до його рецепторів призводить до проліферації ендотеліальних клітин та неоваскуляризації, а також проникності судин, що, як вважається, сприяє розвитку неоваскулярної форми вікової макулярної дегенерації (ВМД), патологічної міопії та хоріоїдальної неоваскуляризації (ХНВ) або порушення зору, спричиненого діабетичним макулярним набряком або макулярним набряком внаслідок тромбозу вен сітківки.
Лікування ексудативної ВМД
Клінічна безпека та ефективність препарату Луцентіс вивчались у трьох рандомізованих, подвійно сліпому та плацебо- або активно-контрольованому дослідженнях з участю пацієнтів з неоваскулярною ВМД тривалістю 24 місяці. Загалом було зареєстровано 1323 пацієнти (879 активної та 444 контрольної груп).
Під час дослідження FVF2598g (MARINA) пацієнти з мінімальними проявами класичної ВМД або прихованими некласичними формами були рандомізовані у співвідношенні 1:1:1 для введення 1 раз на місяць препарату Луцентіс у дозі 0,3 мг і 0,5 мг або плацебо. Загалом було зареєстровано 716 пацієнтів (238 — група плацебо; 238 — Луцентіс, 0,3 мг; 240 — Луцентіс, 0,5 мг).
У ході дослідження FVF2587g (ANCHOR) пацієнти з переважно класичною формою ХНВ були рандомізовані у співвідношенні 1:1:1 для щомісячного введення препарату Луцентіс в дозі 0,3 мг, препарату Луцентіс в дозі 0,5 мг або фотодинамічної терапії (ФДТ) вертепорфіном (на початку дослідження та кожні 3 місяці потому, якщо флуоресцентна ангіографія свідчила про зберігання чи повернення проникності судин). Загалом було зареєстровано 423 пацієнти (143 — група плацебо; 140 — Луцентіс, 0,3 мг; 140 — Луцентіс, 0,5 мг).
Результати обох досліджень свідчать, що подальше лікування ранібізумабом може покращити стан у тих пацієнтів, які втратили ≥ 15 знаків гостроти зору за найкращої корекції (ГЗНК) у перший рік лікування.
Статистично значуще покращення зору за повідомленнями пацієнтів спостерігалося в обох дослідженнях MARINA та ANCHOR під час лікування ранібізумабом порівняно з контрольною групою за опитувальником Національного офтальмологічного інституту (NEI VFQ-25).
Проведено рандомізоване плацебо-контрольоване дослідження FVF3192g (PIER) для оцінки застосування препарату Луцентіс пацієнтам з усіма формами неоваскулярної ВМД.
Пацієнти були рандомізовані у співвідношенні 1:1:1 для ін’єкції препарату Луцентіс у дозах 0,3 мг та 0,5 мг або ін’єкції плацебо 1 раз на місяць протягом 3 місяців поспіль з наступним введенням дози один раз кожні 3 місяці. Загалом у дослідженні було зареєстровано 184 пацієнти. Починаючи з 14-го місяця дослідження, пацієнти, які отримували плацебо, могли переходити на лікування ранібізумабом, а починаючи з 19-го місяця дозволялося більш часте введення препарату. Пацієнти, які отримували лікування препаратом Луцентіс у рамках дослідження PIER, отримали в середньому по 10 ін’єкцій препарату.
Після початкового покращення гостроти зору (після щомісячного введення препарату) в середньому пацієнти, які отримували препарат Луцентіс один раз кожні три місяці, втрачали гостроту зору, повертаючись до базової точки на 12-й місяць. Майже у всіх пацієнтів (82 %), які отримували препарат Луцентіс, гострота зору на 24-й місяць збережена. Дані щодо деяких пацієнтів, які перейшли на лікування ранібізумабом після більш ніж одного року прийому плацебо, свідчать, що ранній початок лікування може асоціюватися з кращим збереженням гостроти зору.
Дані двох досліджень (MONT BLANC, BPD952A2308 та DENALI, BPD952A2309), що завершилися після реєстрації препарату, підтвердили ефективність препарату Луцентіс, але не показують додаткового ефекту комбінованого застосування вертепорфіну (ФДТ препаратом Візудин) та препарату Луцентіс порівняно з монотерапією препаратом Луцентіс.
Лікування порушення зору внаслідок діабетичного макулярного набряку (ДМН)
Ефективність та безпека Луцентісу оцінювалися в ході трьох рандомізованих контрольованих досліджень тривалістю 12 місяців. Усього в цих дослідженнях брали участь 868 пацієнтів (708 — в активній групі та 160 — в контрольній). У ході фази II дослідження D2201 (RESOLVE) 151 пацієнт отримував терапію ранібізумабом (6 мг/мл, n=51, 10 мг/мл, n=51) або плацебо (n=49) у вигляді інтравітреальних ін’єкцій 1 раз на місяць. Середня зміна ГЗНК від 1-го до 12-го місяця порівняно з вихідним рівнем становила +7,8 (±7,72) літери в об’єднаній групі пацієнтів, які отримували лікування ранібізумабом (n=102), порівняно із —0,1 (±9,77) літери у пацієнтів, які отримували плацебо. Середня зміна ГЗНК на 12-му місяці від вихідного рівня становила 10,3 (±9,1) літери порівняно з −1,4 (±14,2) літери відповідно (p < 0,0001 для відмінності між видами лікування).
У ході фази III дослідження D2301 (RESTORE) 345 пацієнтів були рандомізовані у співвідношенні 1:1:1 для отримання або ранібізумабу в дозі 0,5 мг як монотерапії та плацебо-лазерної фотокоагуляції (n=116), комбінованої терапії ранібізумабом в дозі 0,5 мг та лазерної фотокоагуляції (n=118) або ін’єкції плацебо та лазерної фотокоагуляції (n=111).
240 пацієнтів, які до того завершили 12-місячне дослідження RESTORE, були відібрані для участі у відкритому багатоцентровому 24-місячному продовженні дослідження (RESTORE Extension). Пацієнти отримували ін’єкції ранібізумабу 0,5 мг pro re nata (PRN — за потреби) у те ж саме досліджуване око у дослідженні D2301 (RESTORE).
Ефект збігався у більшості підгруп на 12-му місяці. Однак у пацієнтів з достатньо хорошою вихідною ГЗНК (> 73 літер) у разі макулярного набряку з товщиною сітківки в центральній ділянці < 300 мкм не спостерігалося користі від терапії ранібізумабом порівняно з лазерною фотокоагуляцією.
Статистично значуще покращення за повідомленнями пацієнтів стосовно більшості зорових функцій спостерігалося на фоні лікування ранібізумабом (з або без лазерної фотокоагуляції) порівняно з контрольною групою згідно з опитуванням Національного офтальмологічного інституту (NEI VFQ-25). За іншими показниками цього опитування відмінностей між видами лікування не було встановлено.
Профіль безпеки ранібізумабу при довготривалому лікуванні, що спостерігався у 24-місячному розширеному дослідженні, відповідає відомому профілю безпеки препарату Луцентіс.
У дослідженні фази IIIb D2304 (RETAIN) 372 пацієнти були рандомізовані у співвідношенні 1:1:1 для отримання таких препаратів:
- ранібізумаб 0,5 мг з одночасною лазерною фотокоагуляцією в режимі «treat-and-extend» (TE — «лікування та продовження») (n=121);
- ранібізумаб 0,5 мг у вигляді монотерапії в режимі ТЕ (n=128);
- ранібізумаб 0,5 мг у вигляді монотерапії в режимі PRN (n=123).
В усіх групах лікування ранібізумабом починали зі щомісячних ін’єкцій і продовжували, доки ГЗНК не була стабільною щонайменше протягом трьох щомісячних оцінювань поспіль. При TE ранібізумаб застосовували з інтервалами 2−3 місяці. У всіх групах місячна терапія розпочиналася повторно після зниження ГЗНК внаслідок прогресування ДМН та продовжувалася до повторного досягнення стабілізації ГЗНК.
Після перших трьох щомісячних візитів кількість планових лікувальних візитів, передбачених режимом ТЕ, становила 13 порівняно із 20 щомісячними візитами, передбаченими режимом PRN. В умовах застосування обох режимів більше 70 % пацієнтів підтримували свій рівень ГЗНК із частотою візитів ≥ 2 місяці. У дослідженнях ДМН покращення ГЗНК супроводжувалося зменшенням у динаміці середнього показника центральної підпольової товщини сітківки (ЦПТС) в усіх групах лікування.
Лікування порушень зору, зумовлених макулярним набряком, спричиненим тромбозом вен сітківки
Клінічну безпеку та ефективність препарату Луцентіс у пацієнтів із порушеннями зору, зумовленими макулярним набряком, спричиненим тромбозом вен сітківки, вивчали у рандомізованих подвійно сліпих та контрольованих дослідженнях BRAVO та CRUISE з участю пацієнтів із тромбозом гілок вен сітківки (n=397) та тромбозом центральної гілки вен сітківки (n=392). В обох дослідженнях учасникам робили ін’єкції ранібізумабу (0,3 мг або 0,5 мг) або плацебо. Через 6 місяців пацієнтів групи плацебо-контролю переводили на ранібізумаб у дозі 0,5 мг. В обох дослідженнях покращення зору супроводжувалося постійним та значним зменшенням макулярного набряку, на що вказувала товщина сітківки у центрі.
Пацієнти із тромбозом центральної вени сітківки (дослідження CRUISE та розширене дослідження HORIZON): через 2 роки у пацієнтів, які перші 6 місяців отримували плацебо, а в подальшому були переведені на ранібізумаб, покращення гостроти зору (~6 літер) не досягало рівня, подібного до відзначеного у тих, хто приймав ранібізумаб від початку дослідження (~12 літер).
Статистично значуще покращення за повідомленнями пацієнтів за показниками підшкал оцінки ближнього та дальнього зору спостерігалося при лікуванні ранібізумабом порівняно з контрольною групою в опитуванні Національного офтальмологічного інституту (NEI VFQ-25).
Довготривала (24 місяці) клінічна безпека та ефективність препарату Луцентіс у пацієнтів з порушенням зору, зумовленим макулярним набряком, спричиненим тромбозом вен сітківки, оцінювались у ході досліджень BRIGHTER (тромбоз гілок центральної вени сітківки) та CRYSTAL (тромбоз центральної вени сітківки). Під час обох досліджень пацієнтам застосовували режим дозування PRN ранібізумабу в дозі 0,5 мг, що встановлювався за індивідуальними критеріями стабільності. BRIGHTER — рандомізоване активно-контрольоване дослідження в трьох групах для порівняння застосування ранібізумабу в дозі 0,5 мг як монотерапії або в комбінації з ад’ювантною лазерною фотокоагуляцією порівняно з лазерною фотокоагуляцією окремо. Через 6 місяців пацієнти групи лазерної фотокоагуляції могли отримувати 0,5 мг ранібізумабу. CRYSTAL — непорівняльне дослідження застосування ранібізумабу в дозі 0,5 мг як монотерапії. В ході дослідження BRIGHTER ранібізумаб в дозі 0,5 мг з ад’ювантною лазерною терапією продемонстрував не вищу ефективність порівняно із застосуванням ранібізумабу як монотерапії від вихідного рівня до 24-го місяця (95 % ДІ —2,8; 1,4).
Під час обох досліджень швидке та статистично значуще зменшення товщини сітківки в центральній ділянці від вихідного рівня спостерігалося через 1 місяць. Цей ефект зберігався до 24-го місяця.
Ефект лікування ранібізумабом був аналогічним, незважаючи на наявність ішемії сітківки. В ході дослідження BRIGHTER у пацієнтів з ішемією (N=46) або без неї (N=133), які застосовували ранібізумаб як монотерапію, середня зміна від вихідного рівня становила +15,3 та +15,6 літер відповідно на 24-ий місяць.
Під час дослідження CRYSTAL у пацієнтів з ішемією (N=53) або без неї (N=300), які застосовували ранібізумаб як монотерапію, середня зміна від вихідного рівня становила +15,0 та +11,5 літер відповідно.
Під час досліджень BRIGHTER та CRYSTAL покращення зору з часом спостерігалося в усіх пацієнтів, які застосовували 0,5 мг ранібізумабу як монотерапію, незважаючи на тривалість захворювання. У пацієнтів з тривалістю захворювання < 3 місяців покращення гостроти зору на 13,3 та 10,0 літери спостерігалося на 1-ий місяць та на 17,7 та 13,2 літери на 24-ий місяць в ході досліджень BRIGHTER та CRYSTAL відповідно. Відповідне збільшення гостроти зору у пацієнтів з тривалістю захворювання ≥ 12 місяців становило 8,6 та 8,4 літери у вказаних дослідженнях. Слід розглянути можливість початку лікування при діагностуванні захворювання.
Профіль довготривалої безпеки ранібізумабу, що спостерігався у 24-місячних дослідженнях, відповідає відомому профілю безпеки препарату Луцентіс.
Лікування порушення зору внаслідок ХНВ, вторинної відносно патологічної міопії (ПМ)
Клінічну безпеку й ефективність препарату Луцентіс у пацієнтів з порушенням зору внаслідок ХНВ при ПМ оцінювали на підставі даних, отриманих протягом 12 місяців в ході проведення рандомізованого подвійно сліпого контрольованого базового дослідження F2301 (RADIANCE). 277 пацієнтів були рандомізовані у співвідношенні 2:2:1 у такі групи:
• Група I (ранібізумаб по 0,5 мг, режим дозування встановлювався за критеріями «стабільності», які визначалися як відсутність змін у гостроті зору з найкращою корекцією (ГЗНК) порівняно з результатами двох попередніх щомісячних оцінок).
• Група II (ранібізумаб по 0,5 мг, режим дозування встановлювався за критеріями «активності захворювання», які визначалися як порушення зору, пов’язане з інтра- або субретинальною рідиною або активним пропотіванням внаслідок ураження ХНВ, що оцінювалося за допомогою оптичної когерентної томографії (ОКТ) та/або флуоресцентної ангіографії (ФА).
• Група III (ФДТ вертепорфіном, пацієнтам було дозволено одержувати лікування ранібізумабом, починаючи з 3-го місяця).
У групі II, де застосовували рекомендовану дозу препарату, 50,9 % пацієнтів потребували проведення 1 або 2 ін’єкцій, 34,5 % потребували 3−5 ін’єкцій і 14,7 % потребували 6−12 ін’єкцій протягом 12-місячного періоду дослідження. 62,9 % пацієнтів у групі II не потребували ін’єкцій протягом другого 6-місячного періоду дослідження.
Покращення зору супроводжувалося зменшенням центральної товщини сітківки.
Пацієнти в групах застосування ранібізумабу повідомляли про переваги лікування порівняно із застосуванням ФДТ вертепорфіном (р < 0,05) щодо покращення сумарної оцінки та показників деяких підшкал (загальний зір, зір на близькій відстані, психічне здоров’я та залежність) в опитуванні Національного офтальмологічного інституту (NEI VFQ-25).
Діти
Безпека та ефективність застосування ранібізумабу дітям не встановлені.
Європейське агентство з лікарських засобів відклало вимогу щодо надання результатів досліджень препарату Луцентіс в усіх підгрупах педіатричної популяції при неоваскулярній ВМД, порушенні зору внаслідок ДМН, порушенні зору, зумовленому макулярним набряком, спричиненим тромбозом вен сітківки, та порушенні зору внаслідок ХНВ (див. розділ «Спосіб застосування та дози» для отримання інформації про застосування дітям).
Фармакокінетика.
Після щомісячного інтравітреального застосування препарату Луцентіс у пацієнтів з неоваскулярною ВМД концентрація ранібізумабу в сироватці крові була, як правило, низькою, з максимальним рівнем (Cmax), нижчим за необхідну концентрацію, що інгібує біологічну активність судинного ендотеліального фактора росту (VEGF) на 50 % (11−27 нг/мл, як визначено в дослідженні клітинної проліферації in vitro). Cmax була пропорційна до дози в діапазоні доз 0,05−1,0 мг/око. Сироваткові концентрації у деяких пацієнтів з ДМН показують, що не можна виключити дещо вищу системну експозицію порівняно з експозицією у пацієнтів з неоваскулярною ВМД. Сироваткові концентрації ранібізумабу у пацієнтів з тромбозом вен сітківки були практично такими ж або дещо вищими порівняно з відповідними показниками у пацієнтів з неоваскулярною ВМД.
Згідно з даними популяційної фармакокінетики і виведення ранібізумабу із сироватки крові у пацієнтів з неоваскулярною ВМД, які отримували препарат у дозі 0,5 мг, середнє значення періоду напіввиведення ранібізумабу зі склистого тіла становить приблизно 9 днів. При щомісячному інтравітреальному введенні препарату Луцентіс у дозі 0,5 мг в око Cmax ранібізумабу в сироватці спостерігається приблизно через день після введення препарату і становить 0,79−2,90 нг/мл, Cmin може становити 0,07−0,49 нг/мл. Експозиція ранібізумабу у сироватці крові приблизно в 90 000 разів нижча, ніж у склистому тілі.
Фармакокінетика в особливих груп пацієнтів.
Пацієнти з порушенням функції нирок.
Не проводили дослідження фармакокінетики препарату у хворих з порушенням функції нирок. При вивченні фармакокінетики у популяції пацієнтів з неоваскулярною ВМД 68 % (136 із 200) пацієнтів мали порушення функції нирок (46,5 % — незначні [50−80 мл/хв], 20 % — помірні [30−50 мл/хв], 1,5 % — тяжкі [менше 30 мл/хв]). У пацієнтів з тромбозом вен сітківки 48,2 % (253 із 525) мали порушення функції нирок (36,4 % незначне, 9,5 % помірне і 2,3 % тяжке). Рівень системного кліренсу був дещо нижчий, але клінічно незначущий.
Пацієнти з порушенням функції печінки.
Не проводили дослідження фармакокінетики препарату у хворих з порушенням функції печінки.
Клінічні характеристики
Луцентіс Показання
— Лікування неоваскулярної (ексудативної) вікової макулярної дегенерації (ВМД).
— Лікування порушення зору при діабетичному макулярному набряку (ДМН).
— Лікування порушення зору при макулярному набряку, що виник внаслідок тромбозу вен сітківки (тромбоз центральної вени сітківки або тромбоз гілок центральної вени сітківки).
— Лікування порушення зору внаслідок хоріоїдальної неоваскуляризації (ХНВ), вторинної відносно патологічної міопії (ПМ).
Протипоказання
- Підвищена чутливість до активної речовини чи будь-якого іншого інгредієнта препарату.
- Активний/підозрюваний окулярний або періокулярний інфекційний процес.
- Активний тяжкий інтраокулярний запальний процес.
Взаємодія з іншими лікарськими засобами та інші види взаємодій
Досліджень взаємодії не проводилось.
Інформацію щодо ад’юнктивного застосування фотодинамічної терапії вертепорфіном та Луцентісу див. у розділі «Фармакодинаміка».
Інформацію щодо ад’юнктивного застосування лазерної фотокоагуляції та Луцентісу при ДМН та тромбозі гілок центральної вени сітківки див. у розділах «Спосіб застосування та дози»та «Фармакодинаміка».
У клінічних дослідженнях лікування погіршення зору внаслідок ДМН результат стосовно гостроти зору або центральної підпольової товщини сітківки (ЦПТС) у пацієнтів, які отримували лікування препаратом Луцентіс, не залежав від одночасного лікування тіазолідинедіоном.
Особливості застосування
Реакції, пов’язані з інтравітреальною ін’єкцією
Інтравітреальні ін’єкції, включаючи ін’єкції препарату Луцентіс, були асоційовані з ендофтальмітом, інтраокулярним запаленням, регматогенним відшаруванням сітківки ока, розривом сітківки ока та ятрогенною травматичною катарактою (див. розділ «Побічні реакції»). Належна асептична техніка проведення ін’єкцій повинна бути обов’язковою при введенні препарату Луцентіс. Окрім того, потрібно спостерігати за станом пацієнта впродовж тижня після проведення ін’єкції, щоб розпочати своєчасне лікування у разі розвитку інфекційного ускладнення. Пацієнт повинен знати про необхідність негайно повідомляти про появу будь-яких ознак, які можуть нагадувати ендофтальміт або інші вищезазначені ускладнення.
Підвищення внутрішньоочного тиску
Транзиторне підвищення внутрішньоочного тиску відзначали впродовж 60 хвилин після ін’єкції, тому показники внутрішньоочного тиску та перфузії диска зорового нерва слід перевірити і відповідно скоригувати. Також виявляли стійке підвищення внутрішньоочного тиску (див. розділ «Побічні реакції»).
Білатеральна терапія
Деякі дані щодо білатерального застосування препарату Луцентіс (включаючи застосування в один і той же день) свідчать про відсутність підвищеного ризику системних небажаних явищ порівняно з одностороннім введенням.
Імуногенність
Існує ризик виникнення імуногенності при застосуванні препарату Луцентіс. Оскільки у пацієнтів з ДМН можливе збільшення системної експозиції, у цієї популяції пацієнтів існує підвищений ризик розвитку гіперчутливості. Пацієнтів також слід попередити про необхідність повідомляти про збільшення тяжкості внутрішньоочної інфекції, що може бути клінічною ознакою утворення внутрішньоочних антитіл.
Одночасне застосування з іншими препаратами анти- VEGF (судинний ендотеліальний фактор росту)
Не слід одночасно застосовувати Луцентіс з іншими препаратами анти-VEGF (судинний ендотеліальний фактор росту) (при введенні препарату в системний кровообіг або око).
Призупинення застосування препарату Луцентіс
Введення препарату можна припиняти та не поновлювати раніше наступного запланованого введення у разі:
- зниження гостроти зору при найкращій корекції (ГЗНК) на ≥ 30 літер порівняно з попереднім обстеженням гостроти зору;
- внутрішньоочного тиску ≥ 30 мм рт. ст.;
- розриву сітківки;
- субретинального крововиливу, який досягнув центру ямки сітківки, або якщо розмір крововиливу становить ≥ 50 % загальної ураженої ділянки;
- виконаного або запланованого хірургічного втручання на очах протягом 28 днів до або після ін’єкції.
Розрив пігментного епітелію сітківки
Фактори ризику, пов’язані з розривом пігментного епітелію сітківки після застосування інгібіторів VEGF для лікування ексудативної ВМД, включають широке та (або) високе відшаровування пігментного епітелію сітківки. На початку терапії Луцентісом слід бути обережним пацієнтам з факторами ризику розривів пігментного епітелію сітківки.
Регматогенне відшарування сітківки або макулярні отвори
Терапію слід відмінити пацієнтам з регматогенним відшаруванням сітківки або з макулярними отворами 3−4-го ступеня.
Популяції, досвід застосування препарату яким обмежений
Досвід застосування пацієнтам з ДМН внаслідок цукрового діабету І типу обмежений.
Луцентіс не досліджувався при застосуванні пацієнтам, яким раніше вводилися інтравітреальні ін’єкції, пацієнтам з активними системними інфекціями, з проліферативною діабетичною ретинопатією або пацієнтам із супутніми офтальмологічними захворюваннями, такими як відшарування сітківки або макулярні отвори. Також немає досвіду лікування препаратом Луцентіс пацієнтів з діабетом, у яких рівень глікозильованого гемоглобіну HbA1c перевищує норму більш ніж на 12 %, та з неконтрольованою гіпертензією. Лікар повинен врахувати це при лікуванні таких пацієнтів.
Існує недостатньо даних, щоб зробити висновок щодо дії препарату Луцентіс у пацієнтів з тромбозом вен сітківки, в яких спостерігається необоротна втрата зорових функцій внаслідок ішемії.
Існують деякі дані стосовно дії препарату Луцентіс у пацієнтів із ПМ, які не відповіли на попередню фотодинамічну терапію (ФДТ) вертепорфіном. Хоча у пацієнтів із субфовеальними та юкстафовеальними ураженнями спостерігався стійкий ефект, даних для висновку щодо дії препарату Луцентіс у пацієнтів із ПМ та екстрафовеальними ураженнями недостатньо.
Системні ефекти після інтравітреального застосування
Системні небажані явища, в тому числі позаочні крововиливи та артеріальні тромбоемболічні явища, іноді спостерігалися після інтравітреальних ін’єкцій інгібіторів VEGF.
Недостатньо даних щодо безпеки лікування пацієнтів з ДМН, макулярним набряком внаслідок тромбозу вени сітківки та ХНВ, що є вторинною відносно ПМ, а також пацієнтів, які мають в анамнезі інсульт або транзиторні ішемічні атаки. При лікуванні таких пацієнтів необхідно проявляти обережність (див. розділ «Побічні реакції»).
Застосування у період вагітності або годування груддю
Дослідження застосування ранібізумабу у період вагітності не проводили, тому ранібізумаб не слід застосовувати у період вагітності, за винятком випадків, коли потенційна користь перевищує ризик для плода. Дослідження на людиноподібних мавпах не свідчать про прямий або опосередкований несприятливий вплив на перебіг вагітності або розвиток ембріона/плода. Системна експозиція ранібізумабу після очного застосування низька, але з огляду на механізм дії ранібізумаб необхідно розглядати як потенційно тератогенний та ембріо-/фетотоксичний препарат. Жінкам, які планують вагітність та яким вводили ранібізумаб, рекомендовано, щоб між останнім введенням ранібізумабу та зачаттям дитини минуло щонайменше 3 місяці.
Жінкам репродуктивного віку необхідно застосовувати ефективну контрацепцію впродовж лікування.
Невідомо, чи проникає препарат Луцентіс у грудне молоко людини, тому годування груддю не рекомендоване в період лікування препаратом.
Дані щодо фертильності відсутні.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами
Лікування препаратом може спричинити тимчасове порушення зору, що, у свою чергу, може впливати на здатність керувати автотранспортом та іншими механізмами (див. розділ «Побічні реакції»).
Пацієнтам, які відзначали порушення зору, не слід керувати автотранспортом та іншими механізмами до зникнення вищезазначених тимчасових симптомів.
Спосіб застосування Луцентіс та дози
Введення препарату повинен виконувати лише кваліфікований лікар-офтальмолог, який має досвід проведення інтравітреальних ін’єкцій.
Рекомендована доза препарату Луцентіс становить 0,5 мг у вигляді одноразової інтравітреальної ін’єкції. Ця доза відповідає об’єму ін’єкції 0,05 мл. Інтервал між введеннями двох доз в одне око повинен становити щонайменше 4 тижні.
Лікування розпочинати з однієї ін’єкції на місяць і продовжувати, поки не буде досягнута максимальна гострота зору та/або не зникнуть ознаки активності захворювання, тобто зміни гостроти зору та інші симптоми захворювання. Пацієнтам з ексудативною ВМД, ДМН та тромбозом вен сітківки спочатку може бути потрібно три або більше щомісячних ін’єкцій поспіль.
Таким чином, регулярність моніторингу та інтервали між введеннями препарату має визначати лікар залежно від активності захворювання, що оцінюється на основі гостроти зору та/або за анатомічними параметрами.
Якщо, на думку лікаря, показники гостроти зору або анатомічні параметри вказують на відсутність користі від продовження лікування для пацієнта, застосування препарату Луцентіс слід припинити.
Моніторинг активності захворювання може включати клінічне обстеження, функціональні проби або візуалізаційні техніки (наприклад оптичну когерентну томографію або флуоресцентну ангіографію).
Якщо пацієнти отримують лікування за схемою «treat-and-extend» (лікування та продовження), після досягнення максимальної гостроти зору та/або відсутності ознак активності захворювання інтервали між введеннями препарату можна поступово збільшувати до рецидиву ознак активності захворювання або погіршення зору. Інтервал між введеннями препарату слід подовжувати не більше як на два тижні за один раз у випадку ексудативної ВМД, і можна подовжувати максимум на один місяць за один раз у випадку ДМН. У разі тромбозу вен сітківки інтервали між введеннями препарату також можна поступово збільшувати, однак наявні дані є недостатніми, щоб зробити остаточні висновки стосовно тривалості цих інтервалів. У разі рецидиву активності захворювання інтервали між введеннями препарату слід відповідно скоротити.
Лікування порушення зору внаслідок ХНВ слід призначати індивідуально для кожного пацієнта на підставі активності захворювання. Деякі пацієнти можуть потребувати тільки однієї ін’єкції впродовж перших 12 місяців; інші пацієнти можуть потребувати більш частого введення препарату, включаючи щомісячні ін’єкції. Якщо лікування проводиться з приводу ХНВ, що є вторинною відносно патологічної міопії, багато пацієнтів можуть потребувати тільки однієї або двох ін’єкцій протягом першого року (див. розділ «Фармакодинаміка»).
Застосування Л уцентісу та лазерної фотокоагуляції при ДМН та макулярному набряку, спричиненому тромбозом вен сітківки
Був отриманий невеликий досвід введення препарату Луцентіс одночасно з лазерною фотокоагуляцією (див. розділ «Фармакодинаміка»). При застосуванні в один і той самий день Луцентіс потрібно вводити щонайменше через 30 хвилин після лазерної фотокоагуляції. Луцентіс можна застосовувати пацієнтам, яким лазерну фотокоагуляцію проводили раніше.
Застосування Луцентісу та фотодинамічної терапії вертепорфіном при ХНВ, вторинній відносно ПМ
Даних щодо супутнього застосування препарату Луцентіс та вертепорфіну немає.
Інформація про застосування.
Одноразовий флакон тільки для інтравітреального застосування.
Оскільки об’єм, що міститься у флаконі (0,23 мл), перевищує рекомендовану дозу (0,05 мл), частину об’єму, що міститься у флаконі, слід утилізувати до застосування.
Перед застосуванням препарат Луцентіс потрібно візуально перевірити на наявність механічних часток чи зміну кольору.
Введення препарату проводити у стерильних умовах, які включають: дезінфекцію рук як при хірургічному втручанні, стерильні рукавички та серветки, стерильний повікорозширювач (чи еквівалент), стерильний інструмент для парацентезу (якщо потрібно). Слід уважно переглянути алергологічний анамнез пацієнта перед виконанням інтравітреальної ін’єкції (див. розділ «Особливості застосування»). Потрібно продезінфікувати шкіру навколо ока, повіку та поверхню ока. Відповідну анестезію та бактерицидний засіб широкого спектра дії необхідно визначити до проведення ін’єкції згідно з місцевою практикою.
Голку для ін’єкцій потрібно ввести на 3,5—4 мм позаду від лімба у склисте тіло, відхиляючись від горизонтального меридіана і направляючи голку у напрямі до центру очного яблука. Потім ввести 0,05 мл розчину; місце проколу склери потрібно змінювати при подальших ін’єкціях.
Усі компоненти стерильні та призначені тільки для одноразового використання. Будь-який компонент, упаковка якого має пошкодження або ознаки відкриття, не слід застосовувати. Стерильність гарантується тільки за умови цілісності упаковки. Повторне використання може призвести до інфікування або інших захворювань/травм.
Для приготування та інтравітреального введення слід використовувати такі медичні вироби для одноразового застосування:
- фільтрувальна голка на 5 мкм (18G × 1½″, 1,2 мм × 40 мм, у комплекті)
- стерильний шприц 1 мл (включаючи шприц з поділкою 0,05 мл; не входить в упаковку препарату Луцентіс)
- голка для ін’єкцій (30G × ½″; не входить в упаковку препарату Луцентіс).
Під час приготування розчину Луцентіс для інтравітреального введення слід дотримуватися нижченаведеної інструкції.
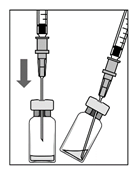
1. Зовнішню частину гумової пробки продезінфікувати перед забором вмісту флакона.
2. З’єднати фільтрувальну голку на 5 мкм (18G × 1½″, 1,2 мм × 40 мм, 5 мкм) зі шприцом 1 мл в асептичних умовах. Натиснути фільтрувальною голкою на центр пробки флакона, поки голка не торкнеться дна флакона.
3. Забрати весь розчин із флакона, тримаючи його вертикально і трішки нахиливши для зручності закінчення забору.
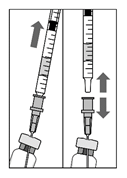
4. Переконатись, що поршень піднятий достатньо, а флакон і голка порожні.
5. Залишити фільтрувальну голку у флаконі, від’єднавши від неї шприц. Фільтрувальну голку слід утилізувати після забору розчину, її не використовують для проведення інтравітреальної ін’єкції.
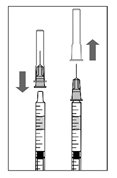
6. В асептичних умовах щільно приєднати голку для ін’єкції (30G × ½″, 0,3 мм × 13 мм) до шприца.
7. Обережно зняти ковпачок голки для ін’єкції так, щоб не роз’єднати голку зі шприцом.
Примітка: знімаючи ковпачок, притримувати за основу голки.
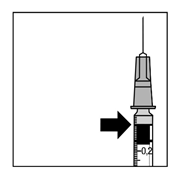
8. Старанно витиснути повітря зі шприца і відрегулювати дозу на рівні 0,05 мл. Шприц готовий для проведення ін’єкції.
Примітка: не протирати голку для ін’єкції. Не тягнути поршень назад.
Після ін’єкції голку не накривати ковпачком знову і не від’єднувати її від шприца. Використаний шприц викинути разом з голкою у контейнер для гострих та ріжучих відходів або утилізувати згідно з місцевими вимогами.
Особливі популяції пацієнтів
Дисфункція печінки
Застосування Луцентісу не досліджували у пацієнтів з порушенням функції печінки. Проте для застосування цій популяції ніякі особливі заходи не потрібні.
Дисфункція нирок
Для пацієнтів з порушенням функції нирок корекція дози не потрібна (див. розділ «Фармакокінетика»).
Пацієнти літнього віку
Пацієнтам літнього віку корекція дози не потрібна. Досвід застосування пацієнтам віком від 75 років з ДМН обмежений.
Діти
Безпека та ефективність застосування ранібізумабу дітям та підліткам (віком до 18 років) не встановлені, тому препарат не застосовують цій групі пацієнтів.
Передозування
Про випадкове передозування повідомлялося під час клінічних досліджень ексудативної ВМД та постмаркетингових досліджень. Побічні реакції, найчастіше пов’язані з передозуванням — це підвищення внутрішньоочного тиску, транзиторна сліпота, зниження гостроти зору, набряк рогівки, біль рогівки та біль в оці. При передозуванні препарату слід перевірити внутрішньоочний тиск та провести відповідне лікування, якщо лікар вважає за потрібне.
Побічні реакції
Більшість побічних реакцій, про які повідомлялося після застосування препарату Луцентіс, пов’язані з процедурою інтравітреальної ін’єкції.
Найчастіші побічні реакції, пов’язані з процедурою введення препарату, включали: біль в оці, гіперемію ока, підвищення внутрішньоочного тиску, запалення склистого тіла, відшарування склистого тіла, крововиливи в сітківку ока, порушення зору, плаваючі помутніння склистого тіла, крововиливи в кон’юнктиву, подразнення ока, відчуття стороннього тіла в оці, підвищене сльозовиділення, блефарит, сухість очей, відчуття свербежу в оці. Найчастіше повідомлялося про такі неофтальмологічні явища, як головний біль, ринофарингіт та артралгія.
Менш часті, але більш тяжкі побічні реакції включали ендофтальміт, сліпоту, відшарування сітківки ока, розрив сітківки ока та ятрогенну травматичну катаракту (див. розділ «Особливості застосування»).
Необхідно попередити пацієнтів щодо можливих побічних реакцій та необхідність звернутися до лікаря у разі розвитку болю в оці, підвищеного дискомфорту в оці, посилення почервоніння ока, нечіткості або погіршення зору, підвищення кількості маленьких часточок у полі зору або збільшення чутливості до світла.
Побічні ефекти, що зазначені нижче, спостерігалися у клінічних дослідженнях. Побічні ефекти# подано за класами систем органів та частотою виникнення згідно з такою систематизацією: дуже поширені (≥ 1/10), поширені (від ≥ 1/100 до < 1/10), непоширені (від ≥ 1/1000 до < 1/100), рідко поширені (від ≥ 1/10000 до < 1/1000), дуже рідко поширені (< 1/10000), невідомої частоти (неможливо оцінити частоту на основі наявних даних). У кожній групі за частотою побічні реакції наводяться у порядку зменшення їх серйозності.
Інфекції та інвазії | |
дуже поширені | ринофарингіт |
поширені | інфекція сечових шляхів* |
З боку крові та лімфатичної системи | |
поширені | анемія |
З боку імунної системи | |
поширені | гіперчутливість |
Психічні розлади | |
поширені | тривожність |
З боку нервової системи | |
дуже поширені | головний біль |
З боку органів зору | |
дуже поширені | запалення склистого тіла, відшарування склистого тіла, крововиливи в сітківку ока, порушення зору, біль в оці, плаваючі помутніння склистого тіла, крововиливи в кон’юнктиву, подразнення ока, відчуття стороннього тіла в оці, підвищене сльозовиділення, блефарит, сухість очей, гіперемія ока, відчуття свербежу в оці |
поширені | дегенерація сітківки, порушення функцій сітківки ока, відшарування сітківки ока, розрив сітківки, відшарування пігментного епітелію сітківки, відрив пігментного епітелію сітківки, зниження гостроти зору, крововиливи в склисте тіло, порушення функції склистого тіла, увеїт, ірит, іридоцикліт, катаракта, субкапсулярна катаракта, помутніння задньої капсули, точковий кератит, ураження рогівки, запалення передньої камери ока, нечіткий зір, геморагія у місці ін’єкції, крововилив в око, кон’юнктивіт, алергічний кон’юнктивіт, виділення з ока, фотопсія, фотофобія, відчуття дискомфорту в оці, набряк повіки, біль у повіці, гіперемія кон’юнктиви |
непоширені | сліпота, ендофтальміт, гіпопіон, гіфема, кератопатія, спайки райдужки, відкладення на рогівці, набряк рогівки, утворення стрій на рогівці, біль у ділянці ін’єкції, подразнення у місці ін’єкції, патологічне відчуття в оці, подразнення повіки |
З боку дихальної системи, органів грудної клітки та середостіння | |
поширені | кашель |
З боку шлунково-кишкового тракту | |
поширені | нудота |
З боку шкіри та підшкірних тканин | |
поширені | алергічні реакції (висипання, кропив’янка, свербіж, еритема) |
З боку скелетно-м’язової системи та сполучної тканини | |
дуже поширені | артралгія |
Обстеження | |
дуже поширені | підвищення внутрішньоочного тиску |
# Побічні ефекти визначали як небажані явища (не менше ніж у 0,5 % пацієнтів), що траплялися з вищою частотою (не менше ніж на 2 %) у пацієнтів, які отримували лікування препаратом Луцентіс, 0,5 мг, порівняно з тими, хто отримував контрольне лікування (плацебо або ФДТ вертепорфіном). * Спостерігалося тільки у пацієнтів з ДМН. | |
Небажані реакції, пов’язані з класом препарату.
У дослідженнях фази III ексудативної ВМД загальна частота неокулярних крововиливів, небажаного явища, потенційно пов’язаного із застосуванням системних інгібіторів VEGF (судинного ендотеліального фактора росту), була трохи вищою у пацієнтів, які отримували ранібізумаб.
Проте закономірності серед різних крововиливів не було. Існує теоретичний ризик появи артеріальних тромбоемболічних ускладнень після інтравітреального введення інгібіторів VEGF, включаючи інсульт та інфаркт міокарда. У клінічних дослідженнях Луцентісу з участю пацієнтів з ВМД, ДМН, тромбозом вен сітківки та ПМ спостерігалася низька частота появи артеріальних тромбоемболічних ускладнень і не було суттєвих відмінностей між групами, які отримували ранібізумаб та контрольний препарат.
Надання повідомлень про розвиток підозрюваних небажаних реакцій.
Надання повідомлень про розвиток підозрюваних небажаних реакцій після реєстрації лікарського засобу є важливим. Це дає змогу вести безперервний моніторинг співвідношення ризик/користь лікарського засобу. Медичних працівників просять повідомляти про розвиток будь-яких підозрюваних небажаних реакцій через національну систему надання повідомлень.
Термін придатності Луцентіс
3 роки.
Умови зберігання Луцентіс
Зберігати у холодильнику (від 2 до 8 °С). Не заморожувати.
Зберігати в оригінальній упаковці та недоступному для дітей місці.
Перед застосуванням закритий флакон може зберігатися при кімнатній температурі (25 °C) протягом до 24 годин.
Несумісність.
Оскільки досліджень несумісності препарату не проводили, цей препарат не можна змішувати з іншими лікарськими засобами.
Упаковка
По 0,23 мл розчину для ін’єкцій у флаконі № 1 у комплекті з голкою в картонній коробці.
Категорія відпуску
За рецептом.
Виробник
Новартіс Фарма Штейн АГ/
Novartis Pharma Stein AG.
Місцезнаходження виробника
Шаффхаусерштрассе, 4332 Штейн, Швейцарія/
Schaffhauserstrasse, 4332 Stein, Switzerland.
Подальша інформація
Пам'ятайте, зберігайте ці та всі інші ліки в недоступному для дітей місці, ніколи не передавайте свої ліки іншим і використовуйте Луцентіс тільки за призначенням.
Завжди консультуйтеся зі своїм лікарем, щоб переконатися, що інформація, яка відображається на цій сторінці, може бути застосована до ваших особистих обставин.
Увага! Ця інструкція для медичного застосування лікарського засобу є офіційною інструкцією виробника Новартіс Фарма Штейн АГ.
Авторське право:
- https://www.novartis.com/ - Новартіс Фарма Штейн АГ
- http://www.drlz.com.ua - Державний реєстр ЛЗ України
| Тип даних | Відомості з реєстру |
| Торгівельне найменування: | Луцентіс |
| Виробник: | Новартіс Фарма Штейн АГ |
| Форма випуску: | розчин для ін'єкцій, 10 мг/мл по 0,23 мл у флаконі; по 1 флакону у комплекті з голкою в картонній коробці |
| Реєстраційне посвідчення: | UA/9924/01/01 |
| Дата початку: | 30.08.2019 |
| Дата закінчення: | 30.08.2024 |
| Міжнародне непатентоване найменування: | Ranibizumab |
| Умови відпуску: | за рецептом |
| Склад: | 1 мл розчину містить 10 мг ранібізумабу |
| Фармакотерапевтична група: | Засоби, що застосовуються при судинних захворюваннях очей. Антинеоваскуляризаційні засоби. |
| Код АТС: | S01LA04 |
| Заявник: | Новартіс Фарма АГ |
| Країна заявника: | Швейцарія |
| Адреса заявника: | Ліхтштрассе 35, 4056 Базель, Швейцарія |
| Тип ЛЗ: | Звичайний |
| ЛЗ біологічного походження: | Так |
| ЛЗ рослинного походження: | Нi |
| Гомеопатичний ЛЗ: | Нi |
| Тип МНН: | Моно |
| Дострокове припинення | Нi |
| Код ATC | Назва групи |
| S | Засоби, що діють на органи чуття |
| S01 | Засоби, що застосовуються в офтальмології |
| S01L | Засоби для застосування при сосудистих очних захворюваннях |
| S01LA | Антинеоваскуляризаційні засоби |
| S01LA04 | Ранібізумаб |